introduction
How are tumor genes detected? Common gene detection technologies include Fluorescence In Situ Hybridization (FISH), Polymerase Chain Reaction (PCR), Sanger sequencing and Next-Generation Sequencing (NGS). NGS is widely used in screening for complex diseases and genetic diseases, tumor assessment, and so on. Tumor Panel testing is based on high-throughput sequencing, featuring a short cycle, high throughput, comprehensive coverage and low cost, which helps with precise diagnosis. Regularly monitoring variations and adjusting treatment plans can provide a basis for clinical trials and off-label drug use. The combination of NGS large panels and other targets is a powerful tool for detecting markers in tumor immunotherapy. However, there are challenges such as incomplete gene coverage and insufficient sequencing depth, which may affect the accuracy of variation detection. Although large panel testing is comprehensive, it is costly, so the pros and cons need to be weighed.
Adaptive sequencing technology, as a unique feature of the nanopore sequencing platform, relies on a sophisticated mechanism controlled by software to achieve efficient enrichment of sequencing sequences. Its core operating principle lies in the fact that, based on the specific sequence information provided by users, it can make intelligent judgments and decide whether to conduct detailed sequencing operations on the target sequences or eject them in a timely manner (as shown in Figure 1). During the operation process, the system will meticulously examine the sequence of the first few hundred bases when DNA molecules pass through the nanopore and accurately compare it with the preset targeted regions. Based on the comparison results, the software will quickly make a judgment: if the nucleic acid molecule exactly meets the requirements of the user’s target sequence, the comprehensive sequencing process will be immediately initiated; on the contrary, if it doesn’t match, the voltage at both ends of the nanopore will be skillfully adjusted to eject the molecule quickly and gracefully, thus freeing up valuable nanopore space for subsequent new nucleic acid molecules. It is particularly worth mentioning that adaptive sequencing technology has greatly simplified the research process. Researchers no longer need to spend a lot of time and energy on tedious preliminary sample preparation or enrichment work and can directly engage in the exciting sequencing process.
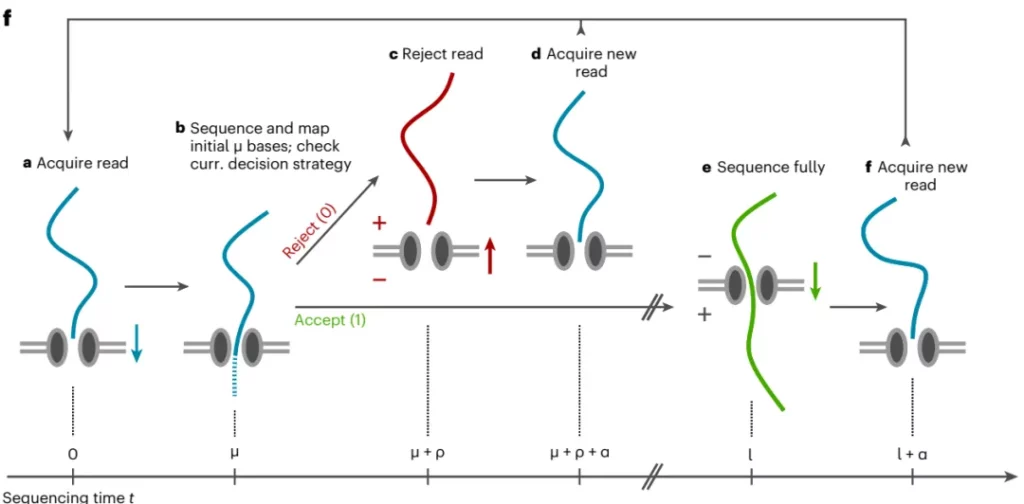
advantages
✔ There is no need for PCR amplification, which reduces amplification bias while retaining base and modification information.
✔ Two modes, enrichment and depletion, are provided. Bioinformatics analysis can easily enrich the target regions without the need for complicated experiments.
✔ The library can be constructed quickly, with the fastest completion time being 10 minutes, which is suitable for rapid clinical applications.
✔ The long-read feature can span large fragment regions, enhancing the ability to detect complex structural variations.
✔ Real-time data analysis can directly enrich the target DNA sequences without additional steps.
✔ It is cost-effective for screening both known and new variations, and is applicable to targeted sequencing of large target regions.
workflow
1. Principle of library construction
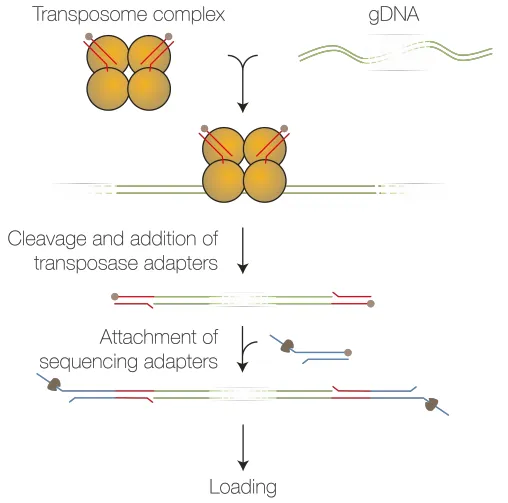
Firstly, extract the genomic DNA samples and fragment the DNA using Tn5. Then, ligate the sequencing adapters to the fragmented DNA. On the Oxford Nanopore Technologies (ONT) sequencing instrument, select the designed bed file targeting specific genes to be detected and conduct the adaptive sampling sequencing procedure.
Research Cases
Case 1
Title: Genome profiling with targeted adaptive sampling long-read sequencing for pediatric leukemia
Journal: Blood Cancer Journal
Impact factor: 12.9
Samples: 28 children with leukemia (10 with acute myeloid leukemia, 13 with B-cell acute lymphoblastic leukemia, and 5 with T-cell ALL)
Technical method: Targeted adaptive sampling long-read sequencing
Targeted sequencing regions: Including 466 genes related to hematological malignancies
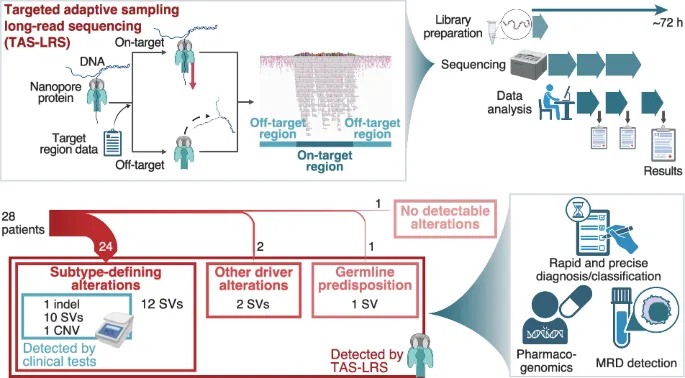
Nanopore sequencers are long-read sequencers with the function of adaptive sampling. They don’t require additional preparation and can cover a wide range of target regions. They perform excellently in structural variation (SV) detection and haplotype variation identification, and the time for library preparation and sequencing is shorter than that of traditional methods. A total of 498 single nucleotide variants (SNVs), 35 small insertions and deletions, and 632 SVs were identified, and some of them were considered as driver changes. In 24 patients, targeted adaptive sampling long-read sequencing (TAS-LRS) determined 85.7% of genomic subtypes and found that all the newly identified variants were SVs. Long-read sequencing combined with targeted capture sequencing can quickly identify multimodal gene variants, including fusion gene SVs, cryptic translocations, and complex SVs. Low-coverage read data can determine copy number variations (CNVs), covering regions as large as an entire chromosome or as small as a few genes. In conclusion, TAS-LRS can rapidly and effectively analyze the genomes of pediatric leukemia through nanopore sequencing, which is beneficial for identifying SVs and CNVs. Its application will improve the practice of leukemia treatment.
Case 2
Title: Assessing the efficacy of target adaptive sampling long-read sequencing through hereditary cancer patient genomes
Journal: Genomic Medicine
Impact factor: 4.7
Samples: 33 patients with suspected hereditary cancer syndromes
Technical method: Targeted adaptive sampling long-read sequencing
Targeted sequencing regions: 147 cancer susceptibility genes
The authors developed a computational workflow for Targeted Adaptive Sampling Long-Read Sequencing (TAS-LRS) and applied it to the genomic assessment of patients with suspected hereditary cancers, identifying single nucleotide variants and clarifying the forms of structural variations. The SVA elements and their origin sites in patients with familial adenomatous polyposis were newly identified. We also demonstrated that the off-target reads of adaptive sampling can be used for SNP genotyping and for calculating polygenic risk scores. Allele-specific hypermethylation of the MLH1 promoter was found in patients with Lynch syndrome. The TAS-LRS workflow can simultaneously capture monogenic and polygenic risk variants as well as epigenetic changes, making it an effective platform for the research and diagnosis of genetic diseases.
Reference
1. Ahsan, M.U., Gouru, A., Chan, J. et al. A signal processing and deep learning framework for methylation detection using Oxford Nanopore sequencing. Nat Commun 15, 1448 (2024). https://doi.org/10.1038/s41467-024-45778-y
2. Kato, S., Sato-Otsubo, A., Nakamura, W. et al. Genome profiling with targeted adaptive sampling long-read sequencing for pediatric leukemia. Blood Cancer J. 14, 145 (2024). https://doi.org/10.1038/s41408-024-01108-5
3. Nakamura, W., Hirata, M., Oda, S. et al. Assessing the efficacy of target adaptive sampling long-read sequencing through hereditary cancer patient genomes. npj Genom. Med. 9, 11 (2024). https://doi.org/10.1038/s41525-024-00394-z
4. Chevrier, Sandy, et al. “Nanopore adaptive sampling detects nucleotide variants and improves large scale rearrangement characterization for diagnosis of cancer predisposition.” Cancer Research 84.6_Supplement (2024): 2936-2936.
https://doi.org/10.1158/1538-7445.AM2024-2936