一、iR IP-seq数据分析方案流程
二、iR IP-seq数据质量分析
Clean reads碱基质量分析 方法描述: 碱基质量值是衡量测序质量的重要指标,碱基质量(Q)与测序错误率(P)密切相关,受测序仪状态,测序试剂质量,样本特性等的影响。质量值计算公式如下:
结果展示
GC含量分析 方法描述 :对测序reads中四种碱基的分布比例进行评估,检查是否存在AT、CG分离现象,理论上A与T、C与G的含量在整个测序反应中分别相同,且维持 在稳定水平。
有效长度统计 方法描述 :去掉index序列、建库平衡用随机碱基及截取掉后面低质量的碱基后,我们用获得的clean reads进行有效长度分析。
cDNA库的代表性分析 方法描述 :如果PCR扩增过度,文库中会含有大量序列完全相同的reads。为了保证文库更好代表原始DNA的丰度,本公司在建库中对PCR扩增循环有严格控制。
PCR duplication level计算方法为 :从测序数据中随机挑选20万reads作为Total Reads,按照如下公式进行计算:
PCR duplication level=Duplication reads/ Total reads
三、iRIP-seq结果展示
全基因组定位分析 Reads比对到参考基因组结果 方法描述 :在这部分分析中,我们将有效测序数据(clean reads)比对到参考基因组上。
Reads在基因组不同区域的分布情况 方法描述 :将clean reads比对到参考基因组上,统计各个区域的分布情况,统计结果如下:
基因组的覆盖度和特征分析 方法描述 :CLIP-seq reads随转录单元长度的覆盖强度分析,以距转录起始位点和转录终止位点为标准,把cDNA平均分成100份,每一份称为一个bin,求落在每个bin中的reads平均数之和,从而得到每个bin上整体的reads覆盖度。
方法描述 :把基因平均分成100份,每一份称为一个bin,求落在每个bin中的reads平均数之和,从而得到每个bin上整体的reads覆盖度。
reads在转录起始位点,转录终止位点,起始密码子和终止密码子附近的分布
方法描述 :分别以转录起始位点/转录终止位点,翻译起始密码子/终止密码子为原点,统计其上下游1kb范围内reads的分布情况。
样本相关性分析 方法描述 :统计落在每个gene上的reads数目,计算每个gene的RPKM值,然后通过比较同一个gene在两个样本中的RPKM值得到两个样本间的相关性。IP 项目中,如果实验与对照样本间相关性系数高,则说明两个样本中大部分reads在染色体上的分布情况类似,暗示实验样本结合的RNA/DNA的富集程度和特异性较低。
样本聚类分析 方法描述 :在这部分分析中,根据样本相关性系数进行样本间聚类分析
四、结合峰分析
结合峰分析策略 方法描述 :因为每个基因的表达量不同,定位在基因组一个位置上的CLIP-seq序列的多少无法评估该RNA结合蛋白在该位置上是否特异性结合。如何剔除基因表达量对RNA结合蛋白特异性结合信号带来的噪音,是分析CLIP-seq数据的一个关键点。MIT的Phillip A Sharp(因发现基因剪接获得过诺贝尔 奖获)实验室2009年一篇论文利用exon array转录本丰度作为参考,来消除表达量对AGO2蛋白所特异结合的mRNA位点的干扰(Chi, Zang et al.
2009)。理论依据是:转录本丰度低的mRNA在IP过程中被RNA结合蛋白抓下来的几率小,反之亦然 。
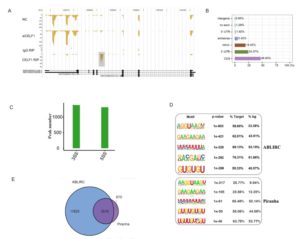
方法一:ABLIRC
我们要求用于分析的单位置比对序列(unique mapped read)彼此之间需要互相重叠,重叠区域至少为1nt。对选定的基因,根据定位到此基因的reads产生随机序列,对此基因进行模拟定位500次,找出500次中最大的随机序列高度(p-value<0.01)作为背景,如果此基因中最大的peak高度< 最大的随机序列高度,则认为该peak为noise,将其去除;反之,则将该peak保留为结合峰。
方法二:Piranha
根据测序深度和覆盖度选取某一固定长度(xx nt)为单位(bin)将基因组等分,统计每个bin中的reads数目。模拟数据中reads的分布情况,来作为背景噪音,然后基于zero truncated negative binomial (ZTNB) 来寻找reads分布显著高于背景的位置。在此过程中每个bin都会获得一个p-value,根据p-value进行显著性筛选,得到真实的结合峰(Uren, Bahrami-Samani et al. 2012)。
结合峰统计 方法描述 :本项目使用上述策略,以input样本为背景,对实验样本的rmdup reads进行Peak Calling,得到实验样本特异的结合峰(peak)并进行统计,结果如下:
结合峰宽度统计 方法描述 :对实验样本特异的结合峰的宽度进行统计,得到结果如下:
结合峰在参考基因组上的分布 方法描述 :统计结合峰在参考基因组上各个区域的分布情况,统计结果如下:
结合峰重叠分析 方法描述 :维恩图主要展示两次重复实验之间重叠的peak的数目。
结合基序分析 方法描述 :我们用HOMER (Hypergeometric Optimization of Motif EnRichment)对实验样本特异的结合峰进行motif分析
结合峰相关基因聚类Gene Ontology 富集分析 方法描述:
1. 利用blast将参考基因组的基因序列比对到Gene Ontology数据库,进行GO注释;
2. 提取比对结果,作为背景即background;
3. 根据结合峰相关基因(peak associated gene)注释信息,统计每个基因所在的GO Term,根据每个Term的基因数目,以及背景中此Term 的基因数目,用超几何分布检验分析每个Term的显著性。
4. 选取排名前10的GO Term及其校正p-value和百分比图进行展示。
结合峰相关基因KEGG Pathway富集分析 方法描述:
1. 利用blast将参考基因组的基因序列比对到KEGG数据库,进行KEGG注释;
2. 提取比对结果,作为背景即background;
3. 根据结合峰相关基因(peak associated gene)注释信息,统计每个基因所在的KEGG pathway,根据每pathway的基因数目,以及背景 中此pathway的基因数目,用超几何分布检验分析每个pathway的显著性。
4. 选取排名前10的pathway及其校正p-value和百分比图进行展示。














 方法描述:CLIP-seq reads随转录单元长度的覆盖强度分析,将基因的5’UTR, CDS, 3’UTR各平均分成100份,每一份称为一个bin,求落在每个bin中的reads 平均数之和,从而得到每个bin上整体的reads覆盖度。
方法描述:CLIP-seq reads随转录单元长度的覆盖强度分析,将基因的5’UTR, CDS, 3’UTR各平均分成100份,每一份称为一个bin,求落在每个bin中的reads 平均数之和,从而得到每个bin上整体的reads覆盖度。