RXBio Translates Sequence to Science and Industry

Traditional single-cell sequencing (such as 10x Genomics 3’ or 5’ end sequencing) can only capture the terminal fragments of transcripts, and has the following limitations:
① Blind spots for isoforms: It is unable to distinguish different splicing variants of the same gene (such as alternative promoters, exon skipping), and these isoforms may have opposite functions (such as oncogenic and tumor-suppressive types).
② Missing complex structural variations: It is difficult to accurately assemble the full-length structures of fusion genes.
The breakthroughs of single-cell third-generation full-length transcriptome sequencing lie in:
✔ Full-length coverage: It can directly read the complete sequence of transcripts from the 5’ cap to the 3’ tail, precisely analyzing splicing sites, UTR variations and RNA editing events.
✔ Single-molecule resolution: It can independently analyze the transcriptome of each cell, avoiding the masking of rare cell types (such as cancer stem cells or immune response cells) due to population averaging.
Full-length transcriptome data provides multi-level information for understanding the regulation of gene expression:
① Diversity of promoters and terminators: The use of different promoters by the same gene can produce proteins with very different functions (such as the pro-apoptotic or pro-survival functions of p53 isoforms).
② Regulatory mechanisms of alternative splicing: By analyzing the activity of splicing factors at the single-cell level, the molecular switches for cell state transitions can be revealed (such as the dynamic changes of Wnt pathway isoforms during embryonic development).
In clinical and translational research, single-cell third-generation full-length transcriptome sequencing is becoming an irreplaceable technology:
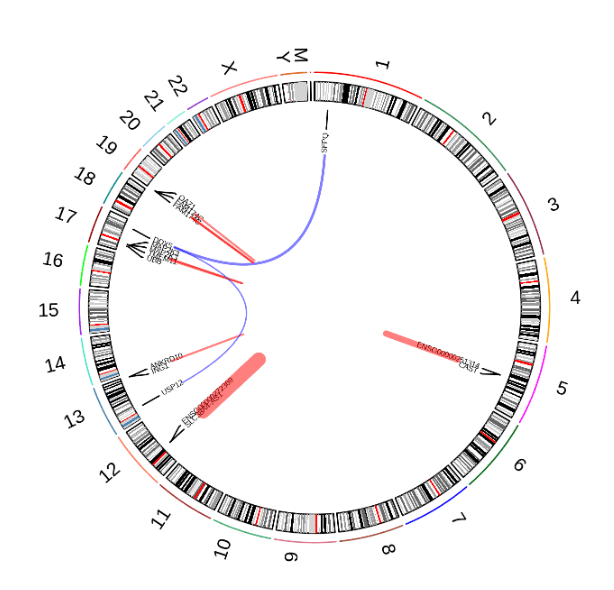
① Analysis of cancer heterogeneity: Identify specific splicing variants that promote metastasis in the tumor microenvironment (such as CD44v6 promoting EMT) and new fusion gene subtypes related to drug resistance (such as BCR-ABL1).
② Diagnosis of rare diseases: Directly detect recessive pathogenic variations caused by splicing errors in single cells (such as exon skipping of SMN1/2 in spinal muscular atrophy).
③ Optimization of immunotherapy: Analyze the full-length diversity of T cell receptors (TCR) to predict the response to immune checkpoint inhibitors.
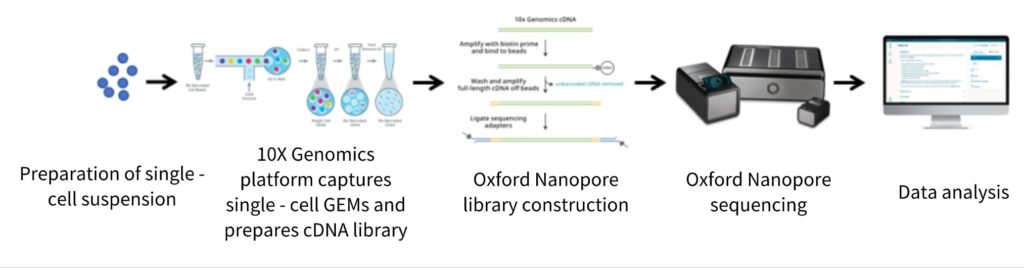
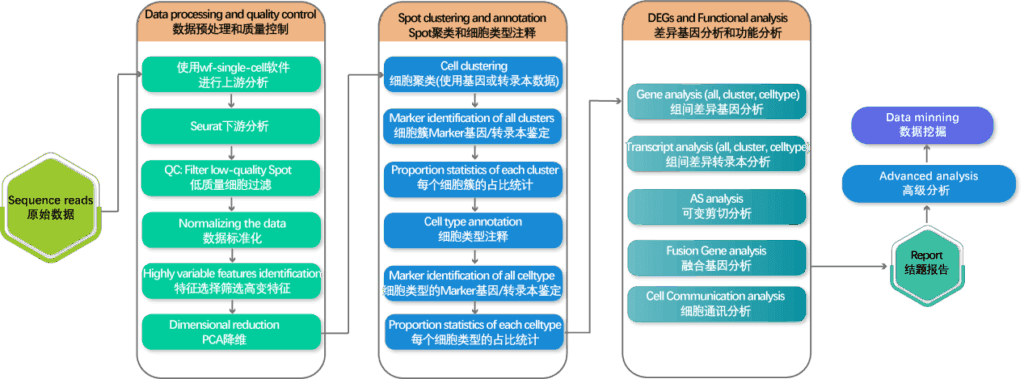
Single-cell third-generation full-length transcriptome sequencing technology is constructed based on the 10x Genomics single-cell capture platform and the Oxford Nanopore long-read sequencing technology. It can not only analyze the panorama of gene expression in individual cells but also accurately capture the complete structural information of mRNA. By integrating the advantages of single-cell technology and third-generation long-read sequencing, this technology advances single-cell transcriptome analysis from the level of gene expression to that of transcript isoforms.
✔ Comprehensive transcript isoform analysis: Accurately analyze all isoform types and provide complete information presentation.
✔ Panoramic analysis of fusion genes: Decode fusion genes in an all-round way and reveal their overall characteristics.
✔ Refined classification of cell subpopulations: Obtain more precise cell subpopulations and improve the accuracy of research results.
✔ Real-time monitoring of alternative splicing: Track change dynamics in real time and control the evolution process.
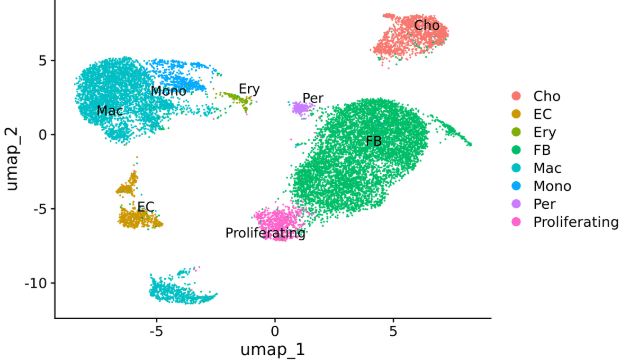
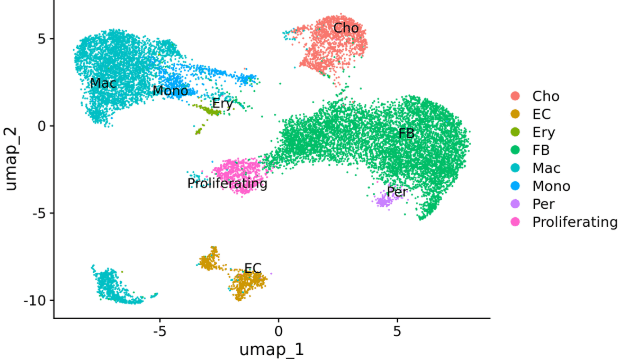
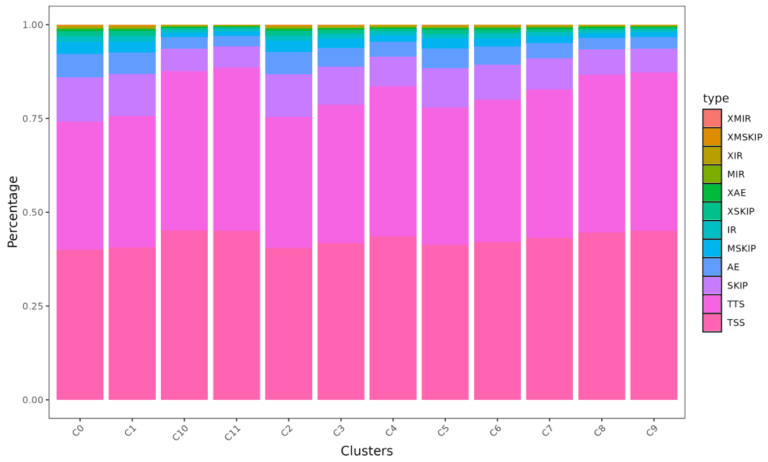
Sample: Mouse lung tissue
Single-cell platform: 10X 3ʹ v3
Third-generation platform: Oxford Nanopore
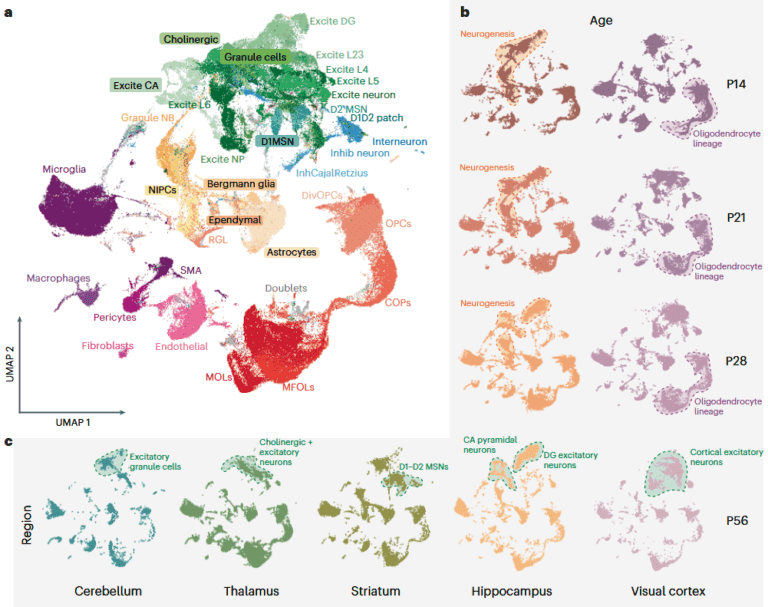
Title:Single-cell long-read sequencing-based mapping reveals specialized splicing patterns in developing and adult mouse and human brain
Journal:Nature Neuroscience
IF:20
Sample information:
Mouse samples covering different developmental stages (P14, P21, P28, and P56) and multiple brain regions (hippocampus, visual cortex, striatum, thalamus, cerebellum) were included. Meanwhile, 6 healthy human brain samples (including the hippocampus, with 3 males and 3 females) were also obtained.
Research findings:
2. Splicing characteristics in regions and developmental stages
3. Splicing conservation and specificity between species
4. Association between splicing and function as well as disease
5. Splicing characteristics of glial cells
Core conclusion:
Splicing diversity is widely present in cell subtypes, brain regions, and developmental stages. It is a key mechanism for shaping the specialization of brain functions and cell identity, and some patterns are conserved between humans and mice. These findings provide a new perspective for understanding the molecular mechanisms of brain development, function, and neurological diseases.