恒河猴脑时空和性别相关lncRNA表达的注释及聚类分析_8-833x1024.png)
RXBio Translates Sequence to Science and Industry

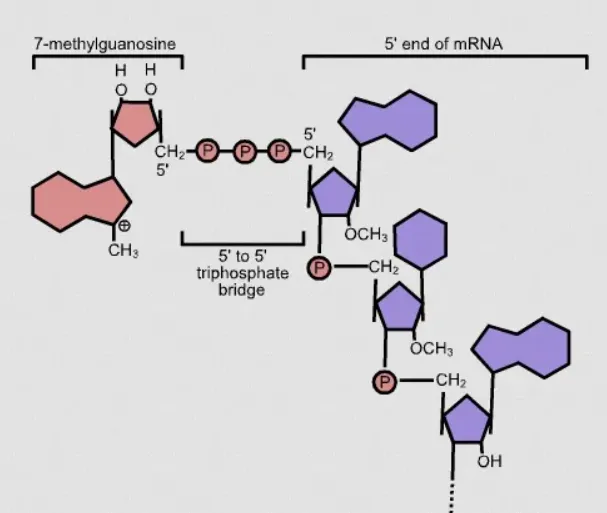
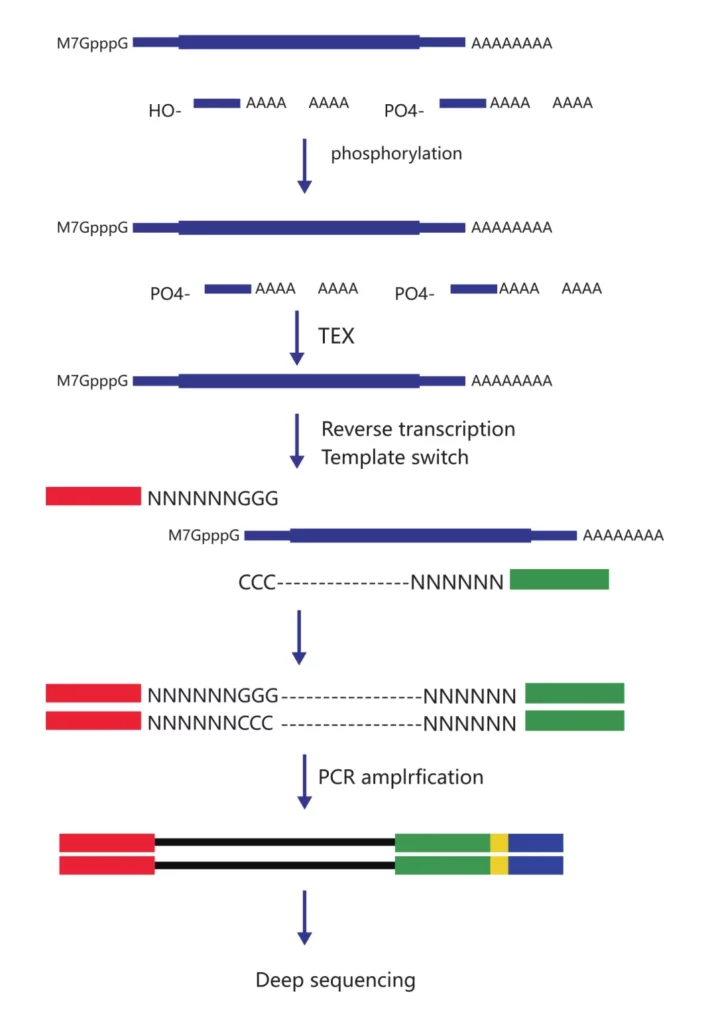
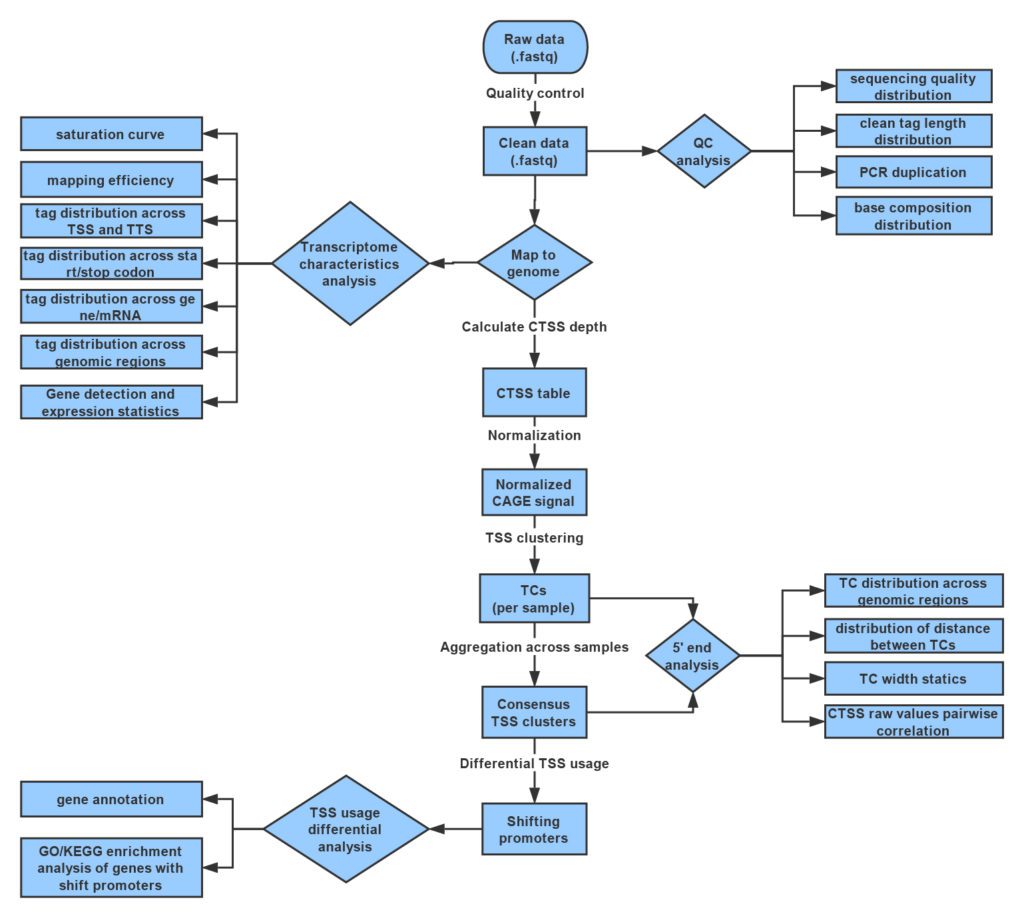
CAGE-seq, namely Cap Analysis of Gene Expression sequencing, is mainly used to identify transcription start sites (TSS). The vast majority of genes have two or even more transcription start sites. Different transcription start sites will cause genes to be regulated by different upstream untranslated regions (5’UTR). Different 5’UTR sequences may contain completely different functional elements, and different start sites result in completely different signals to which gene expression responds. The same gene may be regulated by different promoters, leading to differences in expression and possibly contributing to the occurrence of certain diseases. CAGE-seq can identify all the TSS in mRNA, discover new promoters, conduct differential expression analysis, and also predict possible transcription factor binding sites and the network regulation of gene expression. All of these are achieved through the identification of cap sites.
✔ Provide highly accurate and detailed gene expression analysis by targeting transcription start sites (TSS) rather than the entire gene. It is estimated that there are approximately 50,000 genes in the human genome, but more than 185,000 TSS have been identified. CAGE-seq is capable of recovering most of the TSS.
✔ Quantitative analysis based on each TSS rather than each gene can discover differentially expressed genes that cannot be detected by microarray and RNA-seq.
✔ New genes can be analyzed without the need for DNA chips loaded with probes.
✔ Provide a broader dynamic range to analyze both highly expressed and low expressed genes.
✔ Be able to detect enhancer RNA (eRNA) that is usually expressed at a low level in both directions.
✔ The precise identification of TSS enables better prediction of transcription factor binding motifs than microarray.
On July 7, 2017, the journal Genome Research published an online research paper jointly authored by the research groups of Li Jiali, Hu Xintian, and Zheng Yongtang from the Kunming Institute of Zoology, Chinese Academy of Sciences, and the research group of Wang Xiangting from the School of Life Sciences, University of Science and Technology of China. The study revealed the dynamic changes in expression and the roles of long non-coding RNAs during the development and aging processes of the primate brain.
Title : Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expressiona in Rhesus macaque brain
Journal : Genome Research (Q1, Impact Factor 11.9)
Experimental material: Chinese rhesus macaques
Research Strategy
Using Chinese rhesus macaques as the research subjects, through a series of experimental methods and means such as deep sequencing and analysis of RNA-seq and CAGE-seq, combined with in situ hybridization and functional verification in the primary neuron system, the dynamic change characteristics during the development and aging processes of the Chinese rhesus macaque brain were successfully analyzed.
Research Findings
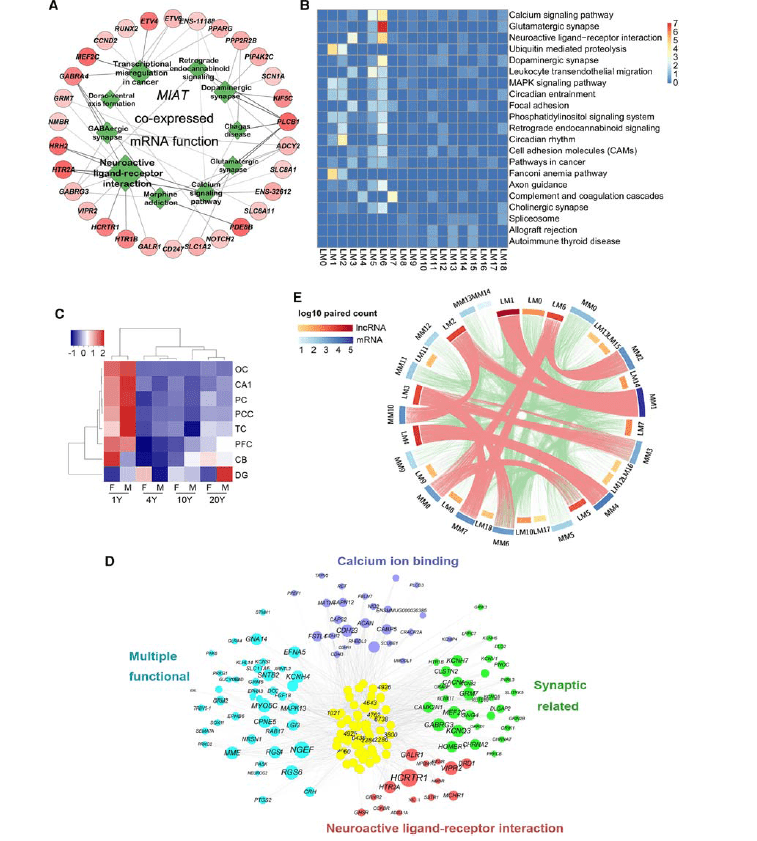
Brain-specific lncRNAs showed a high degree of differences in expression among different regions, genders, and ages.
Based on these expression differences, a total of 18 different lncRNAs and 14 different mRNA expression modules were discovered. Compared with the expression change characteristics of mRNAs, the expression traits of macaque brain-specific lncRNAs exhibited higher dynamic change characteristics during the development and aging processes.
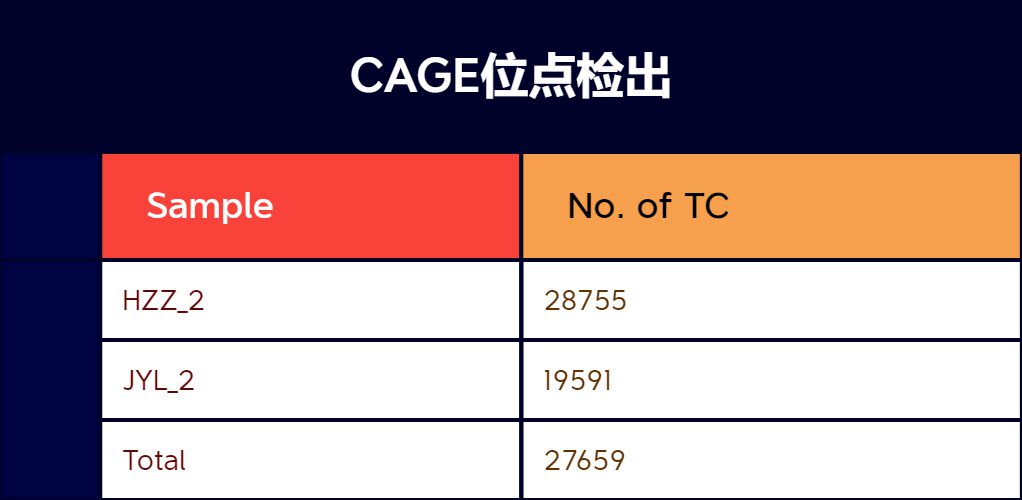
On this basis, CAGE-seq was used to sequence and analyze the promoter regions of lncRNAs and mRNAs. It was found that the changes in the promoter regions of lncRNAs showed more obvious gender differences and spatiotemporal change traits than those of mRNAs.
Further research also found that there were multiple positive and negative regulatory relationships between lncRNAs and mRNAs. Among them, the negative regulatory relationship between lncRNAs and mRNAs specifically expressed in the cerebral cortex region played an important regulatory role in the changes of the structure and neuronal functions of the cerebral cortex during the development and aging processes.
Research Significance
The above research results have provided new experimental evidence for further recognizing and understanding the epigenetic regulatory basis of the complex brain structure and advanced cognitive functions in primates. Moreover, it has also offered new research directions for further exploring the pathogenesis of neurological diseases related to brain development and aging as well as new targets for treatment.