

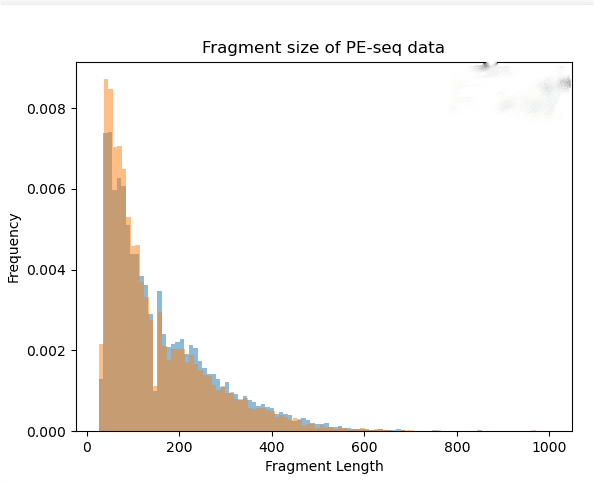
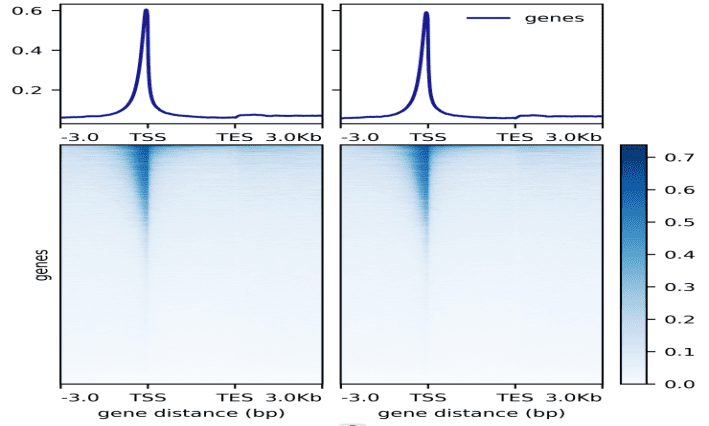
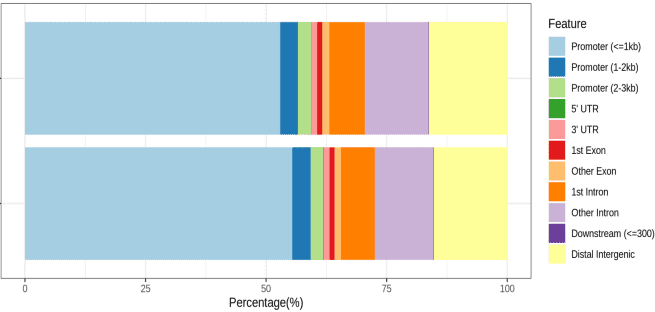
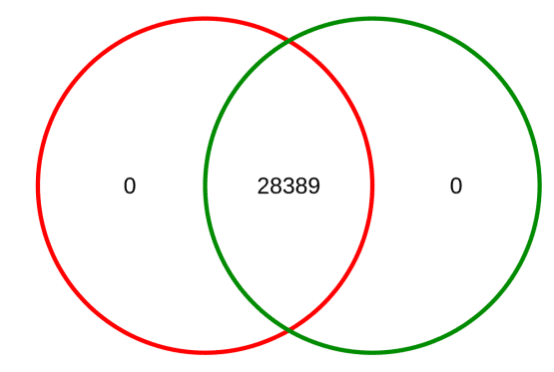
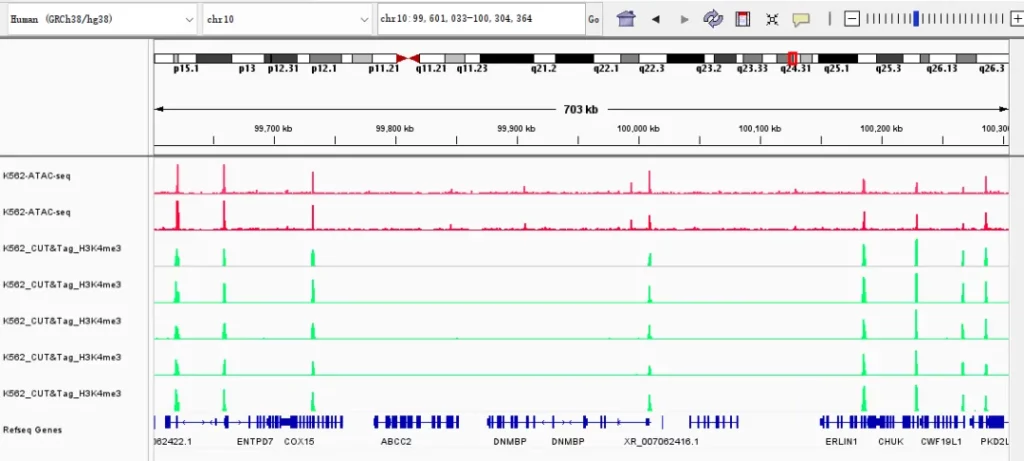
On October 26, 2018, the team led by Professor Howard Y. Chang published the latest achievements in chromatin accessibility research in the international top academic journal “Science”. Using the ATAC-seq method and combining with the existing data from The Cancer Genome Atlas (TCGA) research, the researchers located 562,709 reproducible and transposase-accessible chromatin accessibility sites in 410 frozen samples covering 23 cancer types. In the pan-cancer analysis of chromatin accessibility peaks, approximately two-thirds were consistent with the previously discovered regulatory element information, indicating that the ATAC-seq technology can well reproduce the findings of previous studies and can also discover a large number of new chromatin accessibility sensitive sites at the same time.
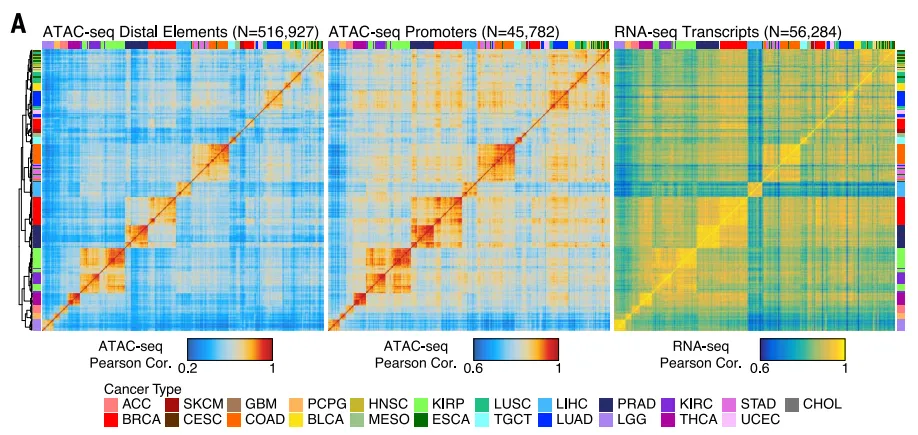
The comparison between the typing based on Distal elements and Promoters data and the typing based on RNA-seq data illustrates that ATAC-seq data can be used to distinguish different tumor types.
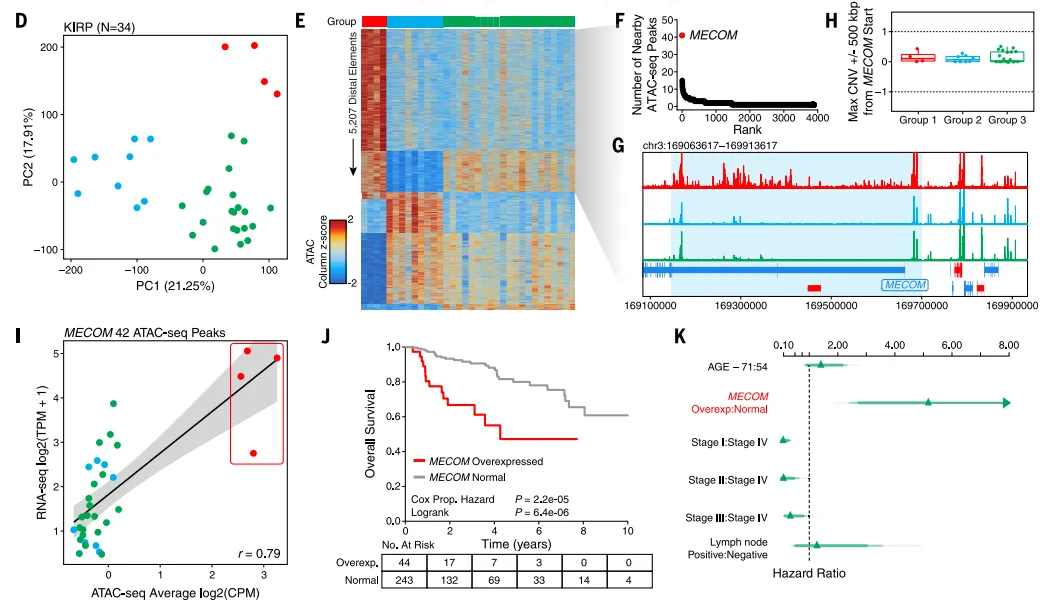
The molecular typing study of KIRP (a type of kidney cancer) using ATAC-seq data found that the red part in the figure represents a new subtype. In this subtype, the chromatin binding activity of the MECOM gene is abnormally increased, which leads to high expression of the MECOM gene. The overexpression of MECOM is significantly associated with poorer overall survival.
Through the search for specific peaks in different types, it was found that these specific peaks are mainly transcription factor binding sites and are closely related to methylation. Moreover, specific transcription factors for different tumor types were also discovered.
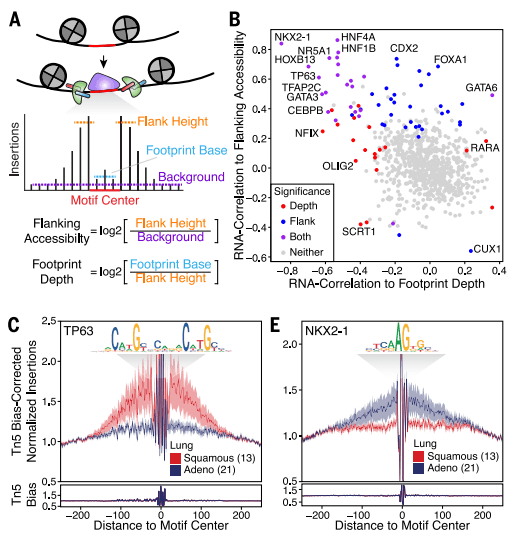
By using ATAC-seq data and RNA-seq data to calculate the correlation between chromatin accessibility and gene expression, a total of 8,552 peaks corresponding to the expression regulation of protein-coding genes were predicted. The specific peaks in different tumor types are different.