
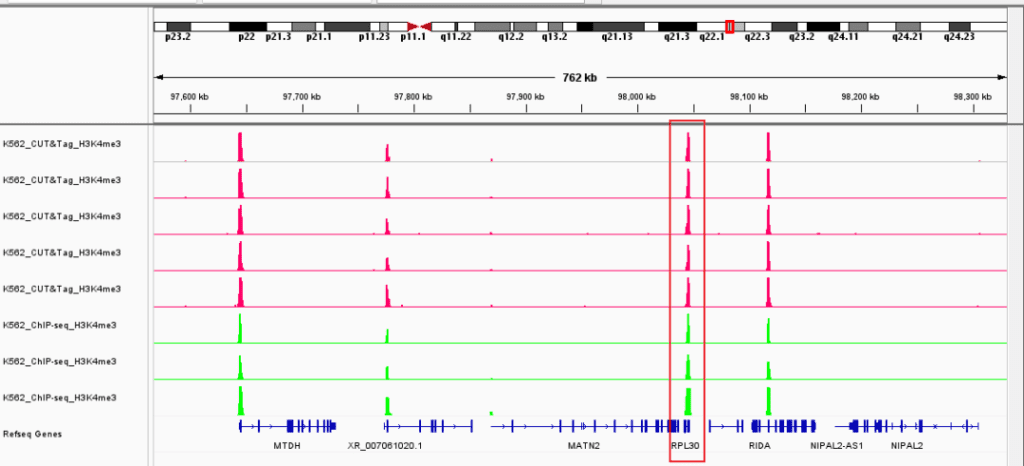

RXBio Translates Sequence to Science and Industry

On April 29, 2019, the Henikoff laboratory announced a revolutionary new technology named CUT&Tag (Cleavage Under Targets and Tagmentation) in Nature Communication. This innovative technology has completely transformed the cell quantity requirement for ChIP-seq experiments, reducing it significantly from the original 10,000 cells to as few as 60 cells or even a single cell, which is undoubtedly a major breakthrough in the field of biological research. The emergence of the CUT&Tag technology has not only greatly improved the feasibility and efficiency of experiments but also achieved a qualitative leap in addressing issues such as the signal-to-noise ratio (the ratio of signal to noise) and data reproducibility that existed in traditional ChIP-seq experiments.
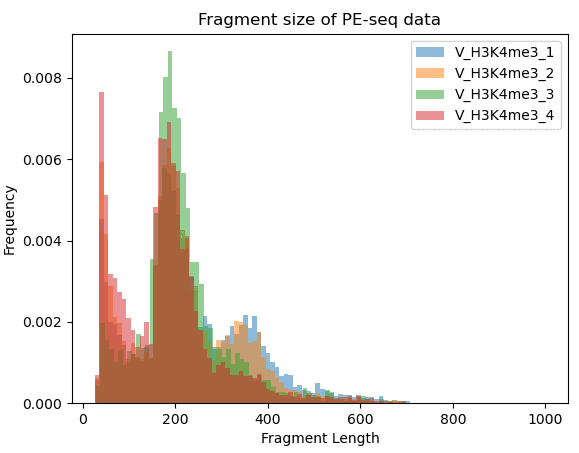
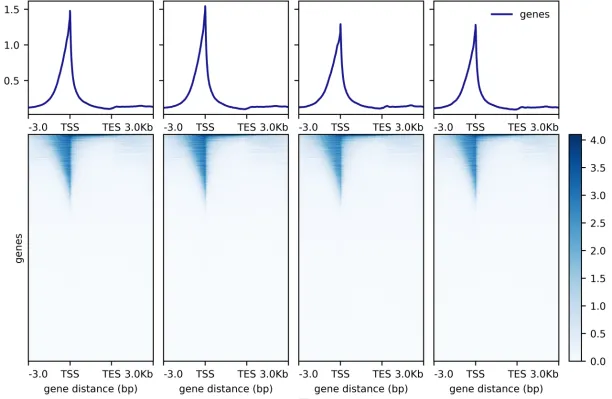
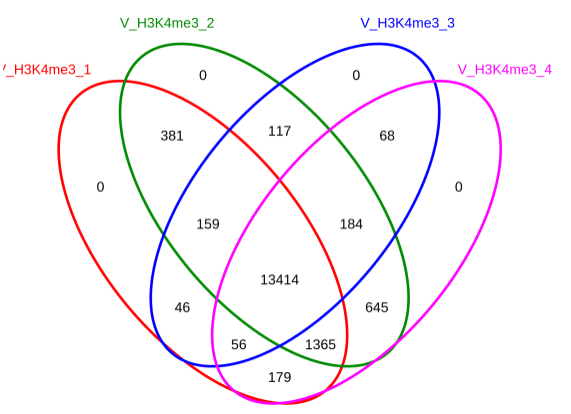
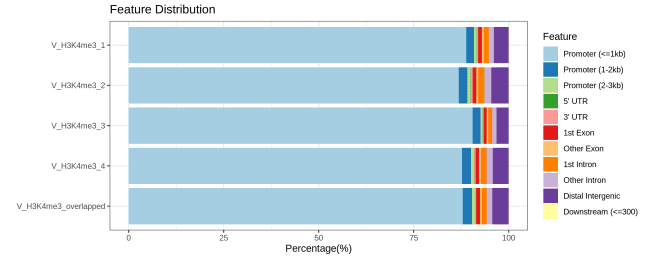
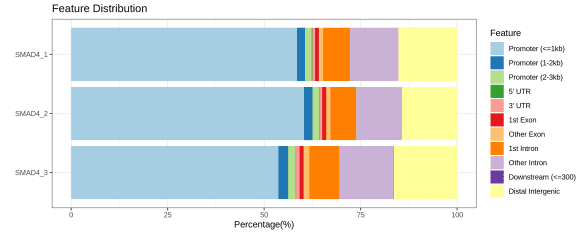
The CUT&Tag technology is an innovative method for studying protein-DNA interactions. It does not require operations such as cross-linking, sonication, end repair, and adapter ligation. Therefore, it has the advantages of requiring a small sample size, having a faster experimental cycle, a high signal-to-noise ratio, and good reproducibility. It can even be used for sequencing at the single-cell level and is especially suitable for research fields such as early embryonic development, stem cells, tumors, and epigenetics. The CUT&Tag is expected to turn the study of protein-DNA interactions into a routine operation similar to a PCR reaction, which is of revolutionary significance for research in fields such as gene regulation and epigenetics.
✔ Low starting amount: The number of cells required ranges from 100 to 100,000 cells, and it can even be used for research at the single-cell level.
✔ Simple workflow: There is no need for formaldehyde cross-linking, sonication, co-immunoprecipitation, end blunting, addition of ‘A’ bases and adapters.
✔ High specificity: It has a higher signal-to-noise ratio and a lower background.
✔ Cost-effective: It requires less sequencing depth.
✔ Good reproducibility: The results of repeated experiments are highly consistent.
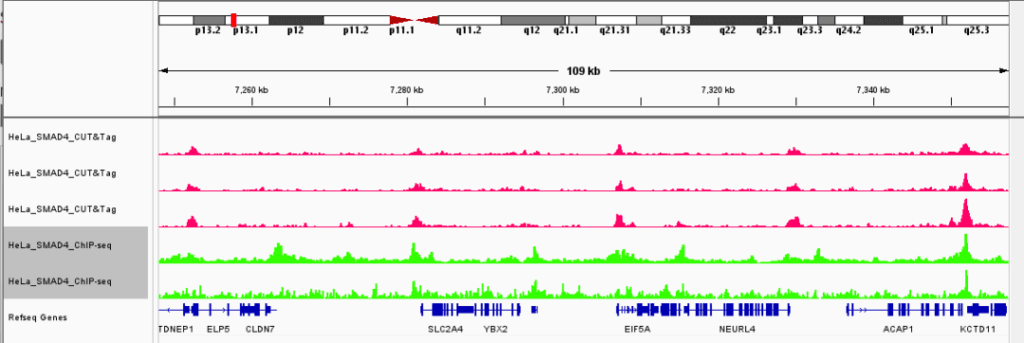
This technology does not require co-immunoprecipitation. Instead, through the mediation of antibodies against the target protein and Protein A, it successfully enables the Tn5 enzyme (which has been fused with Protein A) to add sequencing adapters at both ends of the DNA fragments while cleaving them. After PCR amplification, libraries for high-throughput sequencing can be directly generated. The core step of this technology is “Cleavage Under Targets and Tagmentation”, which conducts genome-wide studies on protein-DNA interactions for extremely small numbers of cells with unprecedented precision and sensitivity. By using the CUT&Tag technology, the amount of input cells and the operation time can be significantly reduced, and high-quality high-throughput sequencing libraries can be obtained in just one day.
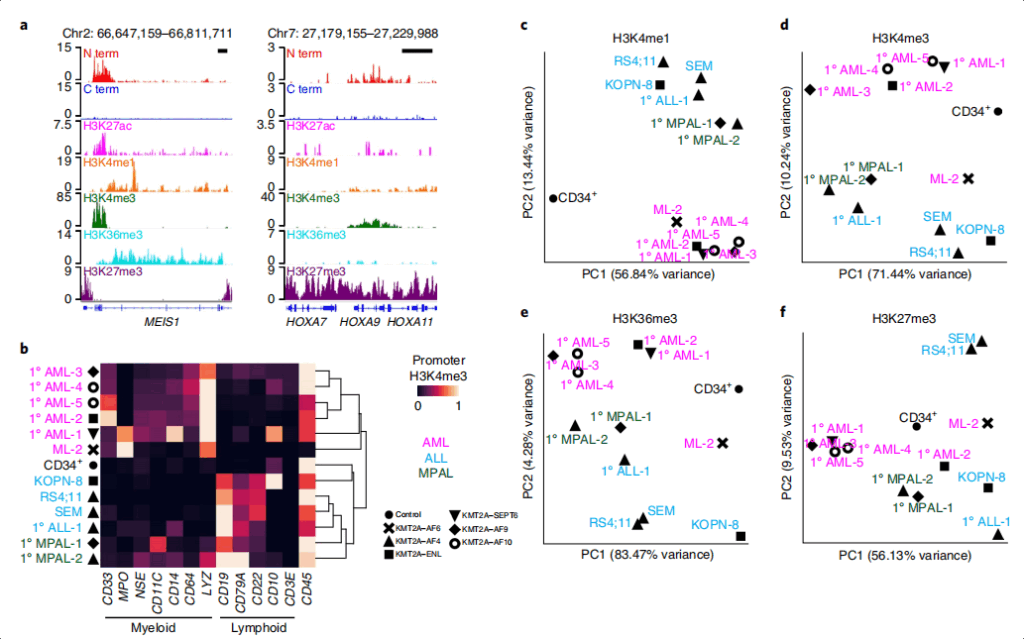
The team led by Steven Henikoff has established the AutoCUT&Tag platform, which can fully automatically analyze tumor fusion proteins, transcription-related complexes and histone modifications in different cell lines and patient samples, thus improving the CUT&Tag chromatin analysis technology. They have identified the specific sites induced by the KMT2A tumor fusion protein and found that different fusion genes exhibit different affinities for various transcription cofactors, which can serve as a tool for predicting the sensitivity of cancers to different therapeutic compounds. The research results have been published in Nature Genetics under the title “Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia”.
The research team utilized AutoCUT&Tag to analyze histone modifications in leukemia samples and discovered the sites of KMT2A tumor fusion proteins with bivalent chromatin characteristics. A subset of the KMT2A tumor fusion binding sites was marked by bivalent (H3K4me3 and H3K27me3) chromatin, and the bivalent chromatin characteristics at these sites were associated with the heterogeneity among the same tumor cells, suggesting that the heterogeneity of gene expression in the mixed-lineage leukemia cell population stems from chromatin dynamics.
Single-cell CUT&Tag analysis of H3K4me3 and H3K27me3 screened out a set of genes shared in most KMT2A-rearranged (KMT2Ar) leukemias. These genes play a crucial role in leukemia cells and are closely related to the growth and differentiation of tumor cells. However, in some KMT2Ar leukemia samples, these target genes are absent, resulting in the greatest changes in active and repressive chromatin marks within the tumor. These missing targets may be due to the binding and activation of tumor proteins in a limited cell subpopulation within the tumor, causing them to drop below the detection level of KMT2A profiling. This also indicates that the KMT2A fusion protein plays an important role in the growth and differentiation of leukemia cells, and this role may be affected by the heterogeneity of gene expression in different cell subpopulations. This finding not only helps to gain a deeper understanding of the role of the KMT2A fusion protein in leukemia but also provides new ideas for the development of targeted therapies against this fusion protein. In future studies, the mechanisms of this gene heterogeneity and how to utilize this information to improve the treatment outcomes of leukemia can be further explored.
Reference:
1. Kaya-Okur, H.S., Wu, S.J., Codomo, C.A. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019). https://doi.org/10.1038/s41467-019-09982-5
2. Janssens, D.H., Meers, M.P., Wu, S.J. et al. Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia. Nat Genet 53, 1586–1596 (2021). https://doi.org/10.1038/s41588-021-00941-9