
Introduction
Whole-genome resequencing is the process of sequencing the genomes of individuals of a species with a known reference genome sequence. By comparing with the reference sequence, it analyzes the genomic differences among individuals or populations. This technology can efficiently and comprehensively detect multiple types of variations in the genome, including single nucleotide polymorphisms (SNP), copy number variations (CNV), insertions/deletions (InDel), and structural variations (SV). With its powerful ability to detect variations, whole-genome resequencing has been widely applied in many fields such as clinical medicine research, population genetics, association analysis, and evolutionary analysis.
The short read lengths (≤300 bp) of second-generation sequencing (NGS) make it difficult to analyze repetitive sequences and complex structural variations. The amplification bias in PCR affects the detection sensitivity of copy number variations (CNV) and other aspects. Third-generation sequencing (LRS), relying on its ultra-long read lengths, can accurately resolve complex regions. Without PCR bias, it improves the uniformity of coverage, and its high accuracy (>99%) supports clinical-level applications. It has significant advantages in aspects such as structural variation and haplotype typing.
ONT whole-genome resequencing (ONT-lrWGRS) is a method for directly sequencing genomic DNA using Oxford Nanopore Technologies (ONT). Its core advantage lies in its ultra-long read lengths, which can effectively span complex repetitive regions and analyze structural variations. Meanwhile, it can detect base modifications (such as methylation). It is suitable for genome assembly, identification of structural variations, and epigenetic research, and has the characteristics of being real-time and fast.
advantages
✔ High variation detection rate: There is no need for PCR amplification, no GC bias, and with ultra-long read lengths, it can easily span highly repetitive and highly complex regions.
✔ Precise variation information: The ultra-long read lengths provide a guarantee for detecting large structural variations (SV).
✔ Richer analysis content: Besides variation information, it can also detect various base modifications on DNA.
workflow
1. Principle of library construction
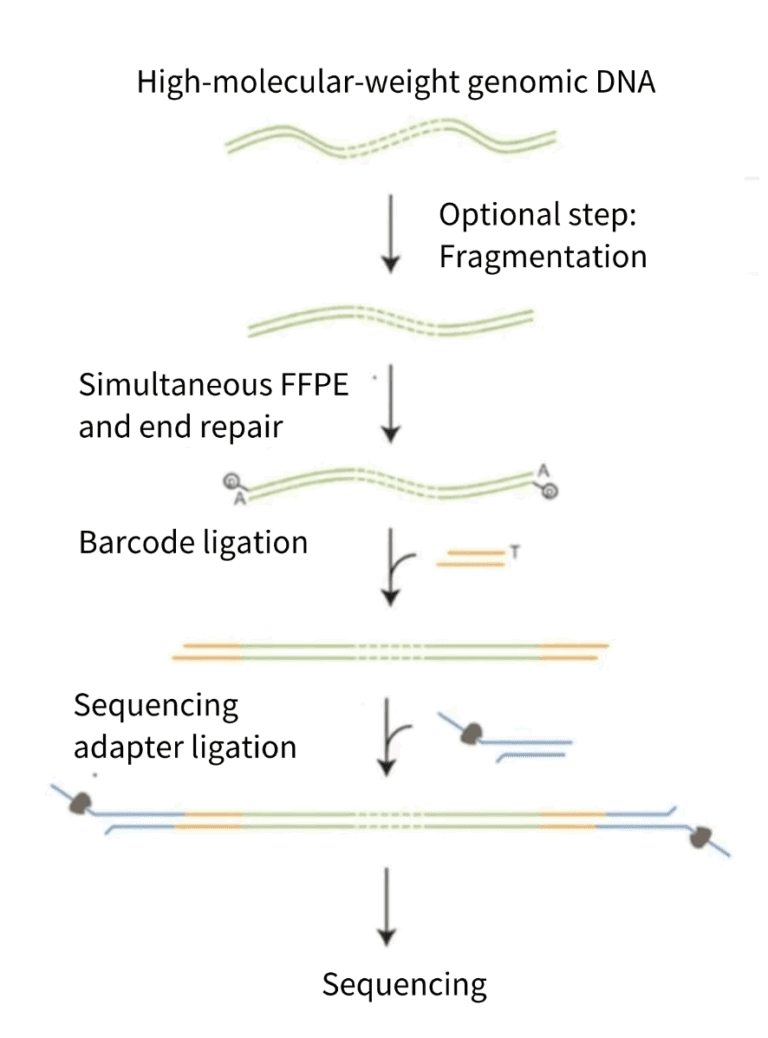
Firstly, high-quality genomic DNA samples are extracted, and then the DNA samples are fragmented (optional). Next, the ends of the DNA samples are repaired and an “A” is added. Subsequently, sample barcodes are ligated, and sequencing adapters are connected. Finally, the samples are loaded onto the sequencing chip for sequencing.
Research Cases
Case 1
Title: Structural variants in the Chinese population and their impact on phenotypes, diseases and population adaptation
Journal:Nature Communications
IF :14.7
Samples: A total of 405 people (206 males and 199 females) were included in this study, with ages ranging from 22 to 81 years old.
Sequencing platform: Oxford Nanopore PromethION
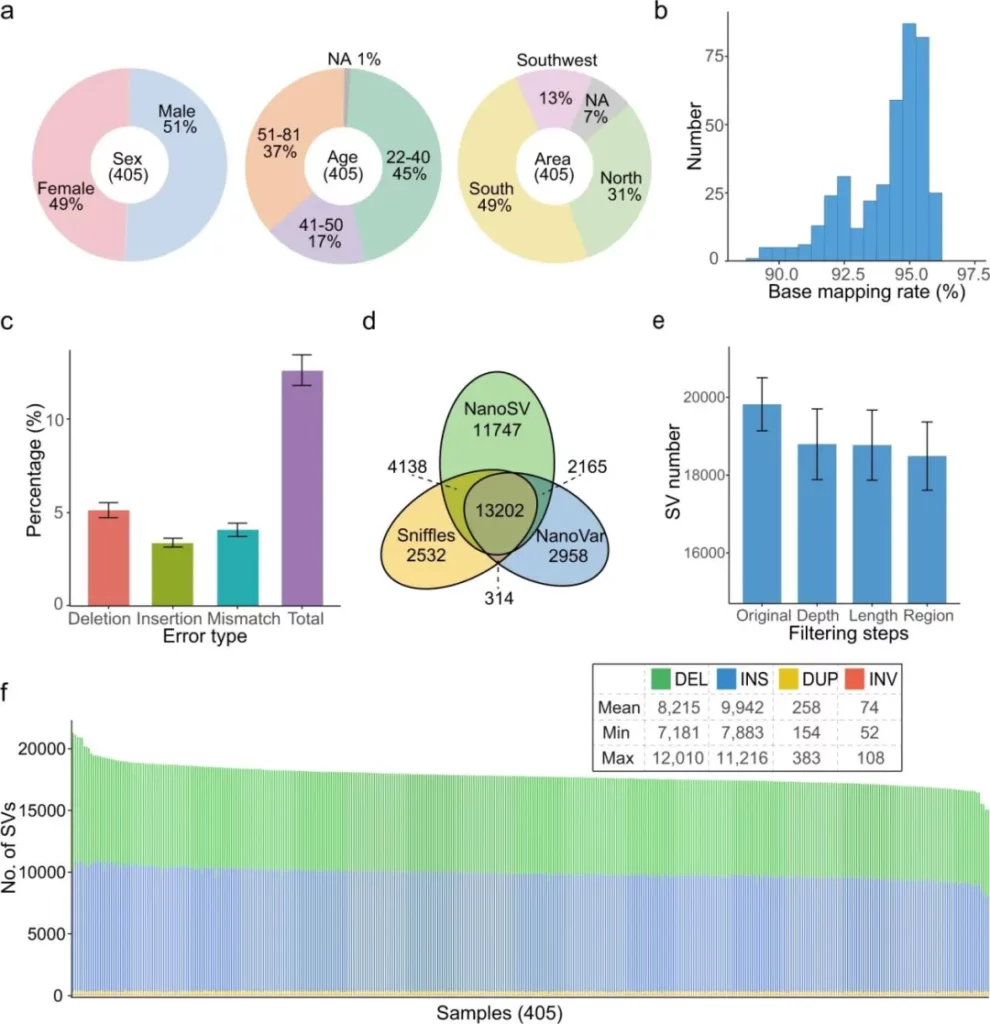
The authors carried out whole-genome long-read sequencing based on the Oxford Nanopore Technologies (ONT) platform. Combined with health screenings, they obtained 68 phenotypic and clinical measurements for 327 individuals and constructed a comprehensive database containing genomic sequencing data and phenotypic/clinical data. After the data underwent quality control, the clean reads were aligned to the reference genome GRCh38, and four types of structural variations (SV) with a length of ≥50 bp were detected, including DEL (deletion), INS (insertion), DUP (duplication), and INV (inversion).
To improve the reliability of SVs, three long-read sequencing genotyping methods, namely Sniffles, NanoVar, and NanoSV, were used for cross-validation. Only the SVs identified by at least two methods were retained, effectively reducing the false positive rate. The results showed that on average, 18,489 high-confidence SVs were identified in each sample, and the number approximated a normal distribution, mainly consisting of DEL and INS. Further analysis found that when the sequencing depth exceeded 15-fold, the growth of the number of SVs tended to saturate, indicating a non-linear association between the sequencing depth and the SV detection rate. The research results provided a quantitative basis for the analysis of the characteristics of gene structural variations and revealed the influence pattern of sequencing depth on the SV detection efficiency.
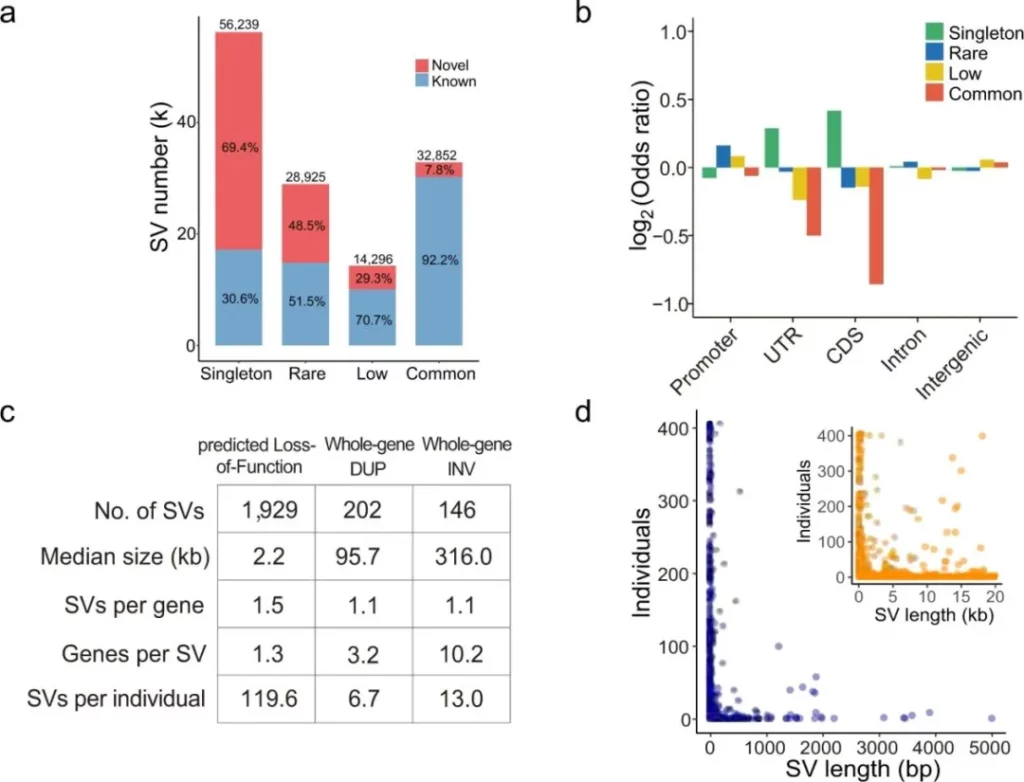
To analyze the potential functions of structural variations (SVs), the authors annotated the SVs based on their genomic positions, covering the coding sequence (CDS) region, untranslated regions (UTRs), promoter regions, and intron regions. The results showed that SVs in the intron region accounted for the highest proportion, and among the SVs in the UTR and CDS regions, singleton SVs (detected only in a single sample) were significantly enriched, suggesting their potential functional tendencies. For SVs that overlapped with the CDS, they were further divided into three subtypes according to the breakpoint positions: predicted loss-of-function (pLoF), which led to the deletion of coding nucleotides or a change in the open reading frame; whole-gene duplication (WDUP), which caused an increase in the overall copy number of the gene; and whole-gene inversion (WINV), which regulated expression by recombining the positions and directions of upstream enhancers and genes. Functional annotation revealed that the 38 genes affected by pLoF SVs were significantly enriched in the “immunoglobulin receptor binding” pathway, implying their potential roles in immune regulation.

To better understand how predicted loss-of-function structural variations (pLoF SVs) affect clinical phenotypes and diseases, the authors annotated the SVs and their related genes using the GWAS catalog, OMIM, and COSMIC databases. For example, a 19.3 kb heterozygous rare deletion (DEL) covers hemoglobin subunit alpha 1 and 2 (HBA 1 and HBA 2), and its dysfunction can lead to alpha-thalassemia; a 27.4 kb heterozygous DEL contains hemoglobin subunit beta (HBB), and its dysfunction can cause severe hemoglobinopathies, such as sickle cell anemia and beta-thalassemia. In addition, the 19.3 kb DEL sequence was found in the gnomAD database, and 92.3% of it came from the East Asian population, indicating that this DEL sequence is population-specific. Finally, a genome-wide association study (GWAS) of clinical phenotypes, such as biochemical, blood, and urine compositions, was further conducted based on genotyped SVs with a minor allele frequency (MAF) > 0.05. The authors also explored the population genetic characteristics between the northern and southern Chinese populations based on deletions (DELs) and insertions (INSs). Principal component analysis (PCA) showed significant genetic diversity between the two groups; the average identity-by-state (IBS) distances among individuals in South China, North China, and between South China and North China were small, indicating that the differences between subgroups were minimal.
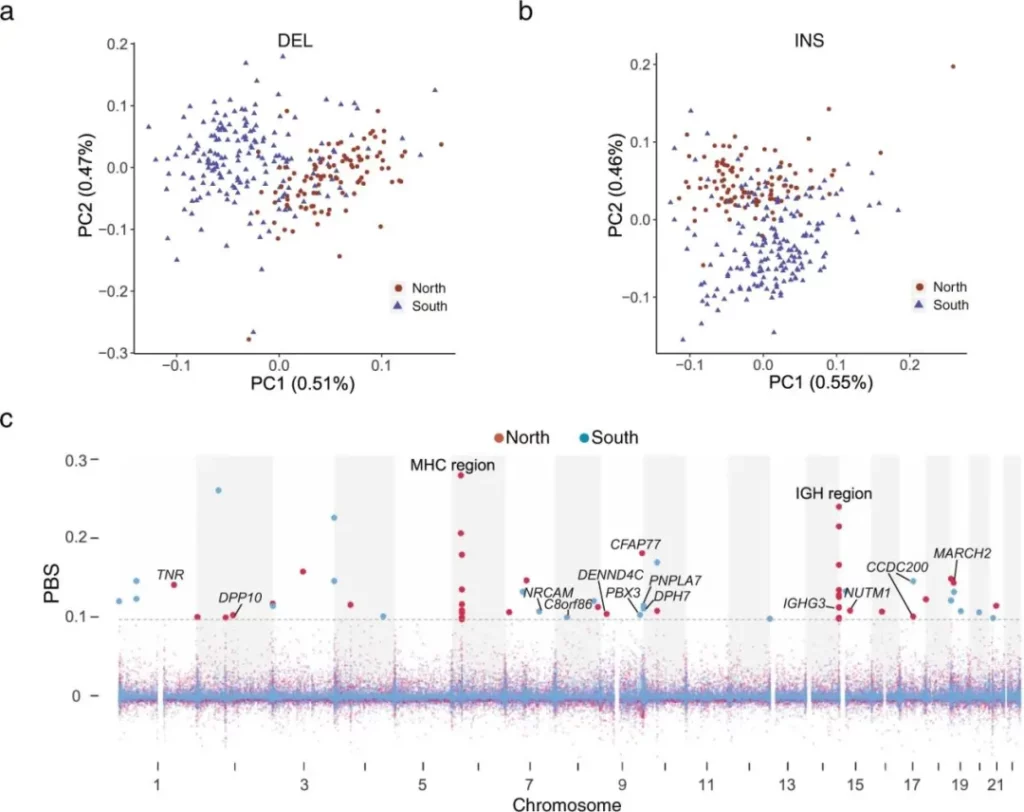
Although previous use of SNP arrays and short-read sequencing (SRS)-based whole-genome sequencing (WGS) has already revealed the genetic differences between individuals in northern and southern China, the authors of this paper observed 24 and 35 independent PBS signals in the genomes of the northern and southern Chinese populations using fixation index (FST) and population branch statistic (PBS), respectively. In the northern Chinese population, the top two signals were located in the major histocompatibility complex (MHC) region (6p21.3 – p22.1) and the immunoglobulin heavy chain (IGH) cluster locus (14q32.33). In the MHC region, nine structural variations (SVs) with PBS signals were located in the intergenic regions of HLA-G, HLA-A, HLA-DRA, HLA-DRB 5, HLA-DRB 1, HLA-DQA 1, and HLADPA 1. Notably, 10 SVs with PBS signals were located in the IGH cluster locus, such as in the intergenic regions of IGHG 3 and TEDC 1, TMEM 121, IGHA 2, IGHE, IGHG 2, IGHA 1, IGHG 1, and IGHG 3, suggesting that the accumulation and combination of different genotypes of IGH genes may be related to immune adaptation to different environments. PBS signals in the MHC and IGH regions were also detected in the southern Chinese population. This result indicates that SVs in immune-related regions may have arisen due to long-term exposure to different environments. In addition, the PBS signal with a 1.4 kb insertion (INS) was located in the 13th intron of PNPLA 7. Based on previous RNA sequencing analysis, PNPLA 7 was associated with hypertension. And the blood pressure and prevalence of hypertension in the northern Chinese population are higher than those in the southern Chinese population. However, more evidence is needed to confirm that the INS in the intron of PNPLA 7 can cause changes in gene expression, thereby leading to changes in blood pressure.
Case 2
Title: Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease
Journal: Nature Genetics
IF: 31.7
Samples: Thirteen patients from 8 families and 4 normal individuals
Sequencing platform: Oxford Nanopore PromethION
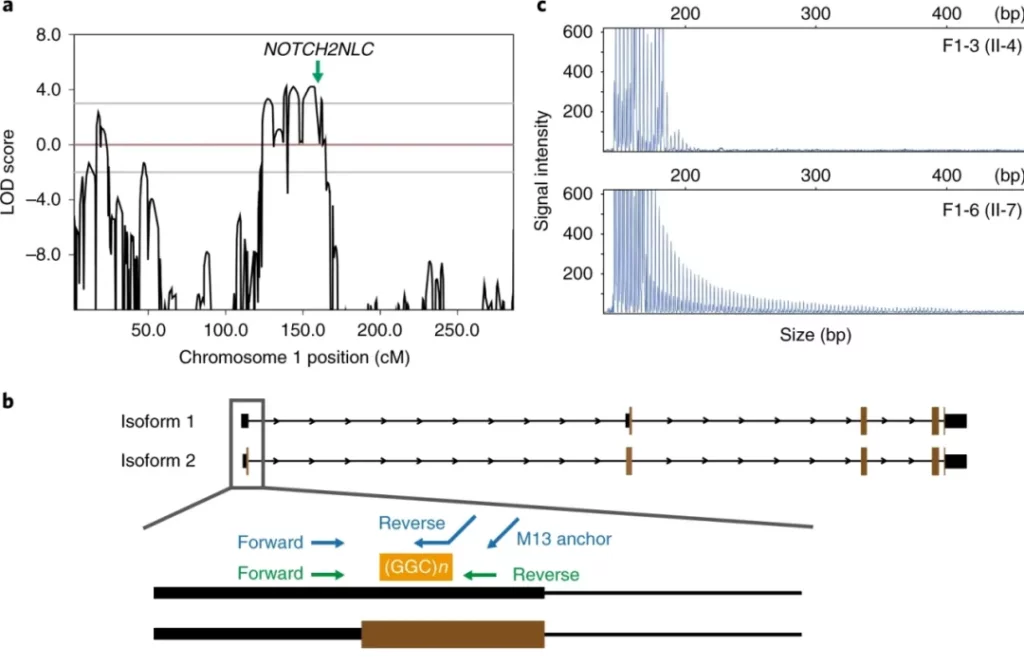
Researchers first conducted second-generation whole-genome sequencing (WGS) or whole-exome sequencing (WES) on affected individuals and normal individuals in family 1. Through linkage analysis, the disease-causing loci were located within an approximately 3.5-Mb region of 1p36.31-p36.22 (chr 1: 6218354-9719813 [maximum LOD 2.32]) and an approximately 58.1-Mb region of 1p22.1-q21.3 (chr 1: 94670784-152323132 [maximum LOD 4.21]). However, no pathogenic SNPs or CNVs were found in the second-generation WGS and WES sequencing data.
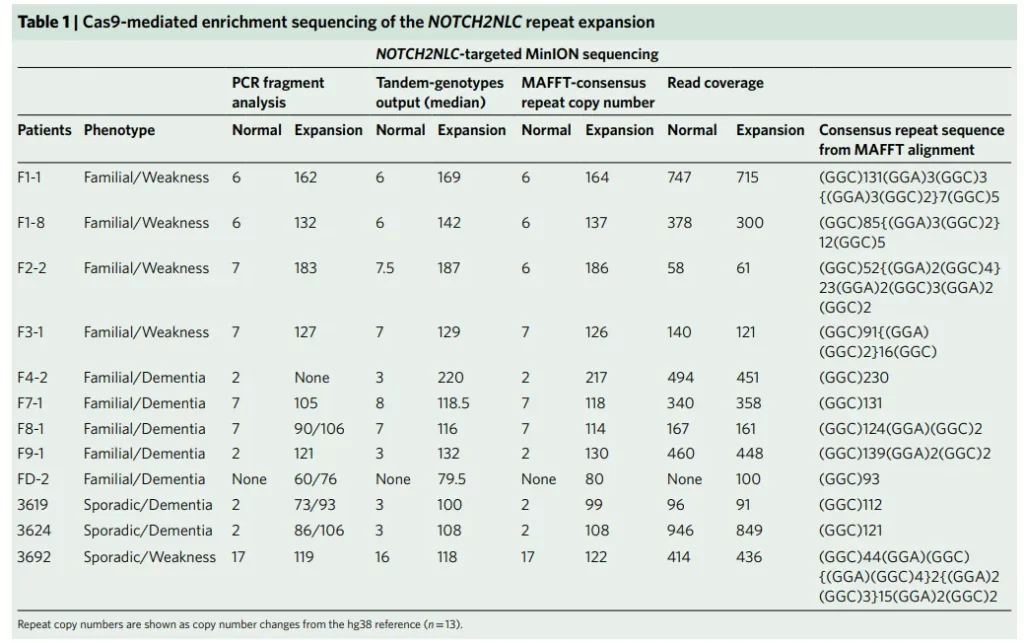
Subsequently, the researchers used the third-generation sequencing platform to conduct whole-genome sequencing on 13 patients from 8 families and 4 normal individuals. Among them, except for patient “F1-6” who was detected using the PacBio RSII platform, the rest of the individuals were detected using the Oxford Nanopore PromethION platform. The sequencing results showed that all 13 patients had GGC repeat expansions at chr1:149390802-149390842. This expanded sequence was located in the 5’UTR region of the NOTCH2NLC gene (NM_001364012) and was positioned within the 58.1-Mb linked region. Further analysis of the repeat expansion region found that in addition to the GGC repeat units, the repeat sequence might also contain GGA repeat units. This was because there were reads supporting the existence of GGA repeat units in the sequencing data of patients from families 1, 2, and 3, and GGA repeat units also existed in the PacBio reads of F1-6.
NOTCH2NLC is located at 1q21.1 and is one of the three human-specific NOTCH2NL genes (NOTCH2NLA, NOTCH2NLB, and NOTCH2NLC). It is highly expressed in a variety of nerve cells, including glial cells, astrocytes, and microglia, and is believed to be involved in the evolution of the human cerebral cortex. The NOTCH2NL genes have a high degree of sequence similarity and are mostly GC-rich regions, so it is difficult to analyze them using methods such as second-generation sequencing. However, long-read sequencing technology has great advantages in analyzing such homologous and repetitive regions. This study also found that when using the Nanopore PromethION platform to conduct whole-genome sequencing on patients with Neuronal Intranuclear Inclusion Disease (NIID), the repeat expansion sequence in the NOTCH2NLC gene could be detected even at a low coverage (~8X).
The researchers also used the Nanopore sequencing data to analyze the methylation level of the GGC repeat expansion sequence and found that there was no difference in the methylation level of NOTCH2NLC between the GGC repeat expansion region and the non-expanded region. Therefore, it is considered that the possibility of CpG methylation caused by GGC expansion leading to the loss of function of NOTCH2NLC is relatively low.
Reference
1.Wu, Z., Jiang, Z., Li, T. et al. Structural variants in the Chinese population and their impact on phenotypes, diseases and population adaptation. Nat Commun 12, 6501 (2021). https://doi.org/10.1038/s41467-021-26856-x
2.Sone, J., Mitsuhashi, S., Fujita, A. et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 51, 1215–1221 (2019). https://doi.org/10.1038/s41588-019-0459-y