
Regulation of Poly(A) Tail and Translation during the Somatic Cell Cycle
V. Narry Kim实验室于2014年开发了可用于高通量检测Poly(A)尾巴长度的TAIL-seq技术,并发现在胚胎细胞以外Poly(A)尾巴长度和翻译效率并无明显的线性相关关系。现在该实验室的研究者们利用TAIL-seq和核糖体分析(ribosome profiling)技术,分析细胞周期中Poly(A)尾巴长度和翻译调控的关系。
We translate sequences to science and industry.

方法描述:碱基质量值是衡量测序质量的重要指标,碱基质量(Q)与测序错误率(P)密切相关,受测序仪状态,测序试剂质量,样本特性等的影响。质量值计算公式如下:


方法描述:对测序reads中四种碱基的分布比例进行评估,检查是否存在AT、CG分离现象,理论上A与T、C与G的含量在整个测序反应中分别相同,且维持 在稳定水平。

方法描述:去掉index序列、建库平衡用随机碱基及截取掉后面低质量的碱基后,我们用获得的clean reads进行有效长度分析。

在cDNA文库构建的过程中对捕获的mRNA/ncRNA 进行随机片段化,随后加接头并进行RT-PCR。一个多样性的文库中大多数序列应该只出现一次,低水平的序列冗余度往往表明高水平的靶标序列覆盖度,而高水平的序列冗余度则意味着一定程度上的偏好富集性,如文库构建过程中PCR过度扩增。通常测序深度越高,越容易产生一定程度的重复reads,属于正常的现象。实际操作中,由于数据量较大,为了降低计算中对内存的要求,仅选取了每个文件的前200,000条reads进行分析,认为其可以代表全部序列的冗余度。
PCR duplication level计算方法为:从测序数据中随机挑选20万reads作为Total Reads,按照如下公式进行计算:PCR duplication level=Duplication Reads/Total Reads

方法描述:根据不同的基因组的特征,选取相对合适的软件,动植物用HISAT2 (Kim D, Langmead B et al. 2015)、真菌或者基因密度较高的物种用Bowtie2(Langmead and Salzberg 2012),根据需要会设定一定的容错率,将有效测序数据(clean reads)比对到参考基因组上。

方法描述:统计在基因组上有唯一定位的reads在各个区域的分布情况

方法描述:reads随转录单元长度的覆盖强度分析,以距转录起始位点和转录终止位点为标准,把cDNA平均分成100份,每一份称为一个bin,求落在每个bin中reads平均数之和,从而得到每个bin上整体的reads覆盖度
 Reads在转录起始位点,转录终止位点,起始密码子和终止密码子附近的分布
Reads在转录起始位点,转录终止位点,起始密码子和终止密码子附近的分布方法描述:分别以转录起始位点(TSS)和转录终止位点(TTS)为原点,统计其上下游1kb范围内reads的分布情况,结果如下:

方法描述:分别以起始密码子(start codon)和终止密码子(stop codon)为原点,统计其上下游1kb范围内reads的分布情况,结果如下:

方法描述:RPKM (Reads per kilo base of a gene per million reads),表示每百万 reads 中来自于某基因每千碱基长度的 reads 数,表征基因的表达丰度(Mortazavi, Williams et al. 2008)。通过对mRNA长度和测序深度进行均一化(RPKM),使不同测序样本之间的表达丰度具有可比性,消除了因mRNA长度和不同样本之间测序深度差异可能导致的偏差,RPKM计算公式:RPKM=total exon reads/(uniquely mapped reads*exon length)

方法描述:在文库构建中,加入了用于对照的长度为120nt和40nt的poly(A) tail spike-in序列,统计测序数据中检出的spike-in序列中poly(A)的长度分布,结果如下:

方法描述:对每个样本中含有不同长度poly(A)的polyA_clean_reads的数目进行统计,结果如下:

方法描述:对每个样本中的polyA_clean_reads,将截掉A之后剩余部分的reads比对到参考基因组上,统计比对上的这些reads所在基因的poly(A)长度的中位数,然后得到每个样本中具有不同poly(A)长度中位数的基因的数目,分布如下:

方法描述:对于不同处理的样本,使用秩和检验(Rakumtet)分析poly(A)长度的中位数的差异情况,筛选poly(A)长度中位数变化显著(满足p-value <=0.01)的基因,并根据poly(A)长度中位数差异的绝对值的不同范围,分成1-10nt,11-20nt,21-30nt及30nt以上等组,进一步统计不同poly(A)长度变化范围的基因数目。



V. Narry Kim实验室于2014年开发了可用于高通量检测Poly(A)尾巴长度的TAIL-seq技术,并发现在胚胎细胞以外Poly(A)尾巴长度和翻译效率并无明显的线性相关关系。现在该实验室的研究者们利用TAIL-seq和核糖体分析(ribosome profiling)技术,分析细胞周期中Poly(A)尾巴长度和翻译调控的关系。
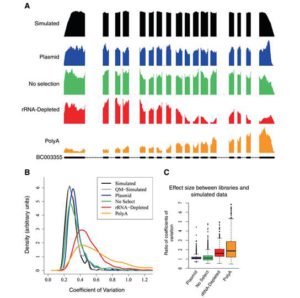
宾夕法尼亚大学的研究者们最近使用了一项新技术来检测在RNA-seq建库和测序过程中人为引入的偏好。高通量RNA测序技术已称为基因表达和转录组分析的重要手段,但人们对不同的建库方法引入的偏好(bias)了解不足,迄今为止也没有好的手段来估算这种偏好。John B. Hogenesch的研究团队展示了通过体外转录组测序(in vitro transcription seq,IVT-seq)估算偏差的方法,可能为今后的实验设计和方法改进提供参考。
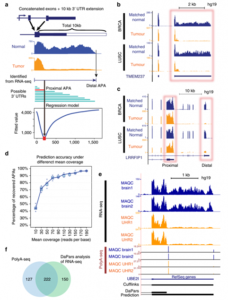
可变聚腺苷酸化(alternative polyadenylation, APA)是转录后水平调控的重要组成部分。约70%的人类基因都有多个polyA位点,可以产生长度不同的3′-UTR或transcript variant,极大提高了转录组的多样性。最近的研究显示肿瘤组织也有其独特的APA特征,如形成较短的3′-UTR,但人们对其的认识还远远不够充分。贝勒医学院Wei Li研究团队在Nature Communication上报道使用一种新的生物信息学算法DaPars(Dynamic analysis of Alternative PolyAdenylation from RNA-Seq)重新分析了现有的肿瘤组织RNA-seq数据,他们发现的肿瘤组织APA特征可能为癌症诊断提供新的方法。
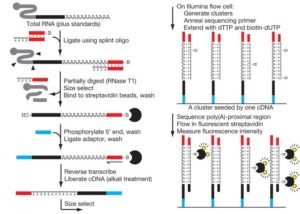
Subtelny等研究人员发表了题为“Poly(A)-tail profiling reveals an embryonic switch in translational control poly(A)”的文章,通过PAL-seq (poly(A)-tail profiling by sequencing) 技术研究了胚胎发育时期poly(A)尾巴长度与翻译效率的关系。相关成果公布在2014年1月29日Nature杂志上。