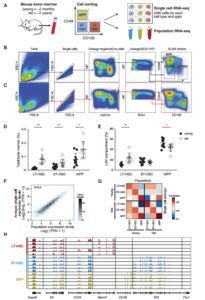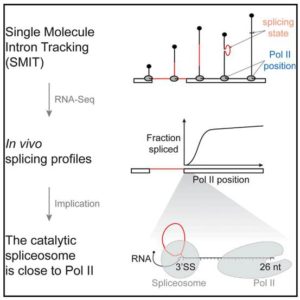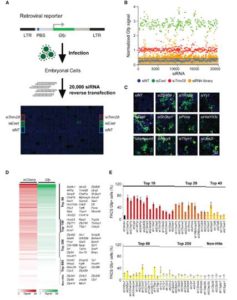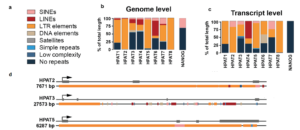
Argonaute CLIP Defines a Deregulated miR-122-Bound Transcriptome that Correlates with Patient Survival in Human Liver Cancer
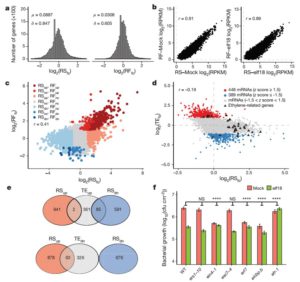
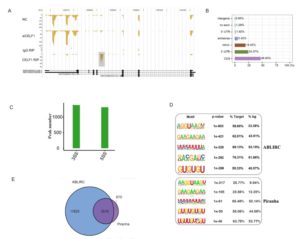
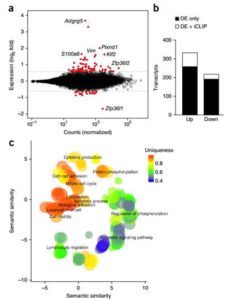
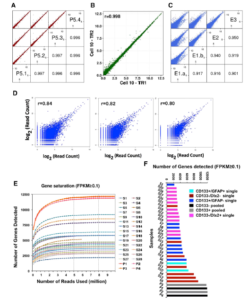
miR-122是一个肝脏组织特异表达的miRNA,长度在21-22 nt。miR-122通过和Ago蛋白的结合,会进一步结合到靶标基因的3’UTR上面,从而造成基因的转录后调控,会造成数百个基因的表达抑制。miR-122在肝脏中的功能研究已经比较多,尤其是在肝癌发生过程中的研究。miR-122的敲除并不会太大影响肝脏的发育和功能,但会造成年轻人的肝脏炎症反应以及脂肪肝,在年老人群中,会进一步引起肝癌。在肝癌病人中,前人也发现了miR-122的低表达。综合上述结果,miR-122被认为是一个抑癌因子,而且可以进一步作为肝癌治疗的一种药物靶点。但是到目前为止,miR-122在肝脏中的调控网络,以及作用的靶基因之间的关系还不是很完善。