
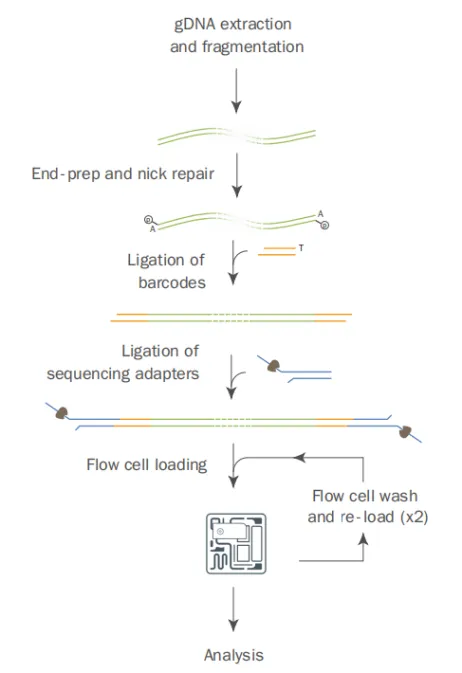
The research team from the University of Pennsylvania in the United States has developed an analysis pipeline for long-read genomic DNA methylation adaptive sampling sequencing. The research results were published in Nature Communications, and the article was titled “A signal processing and deep learning framework for methylation detection using Oxford Nanopore sequencing”.

Oxford Nanopore sequencing can detect DNA methylation and has unique advantages. Adaptive sampling reduced representation methylation sequencing is applicable to CpG islands or imprinted regions. DeepMod2 is a deep learning framework that utilizes the ionic current signals of nanopore sequencing to detect DNA methylation. DeepMod2 runs efficiently on CPUs and can infer epigenetic haplotypes or methylation calls. Compared with the closed-source tools Guppy and Dorado, DeepMod2 has comparable performance. In conclusion, DeepMod2 is a fast and accurate open-source DNA methylation detection tool.
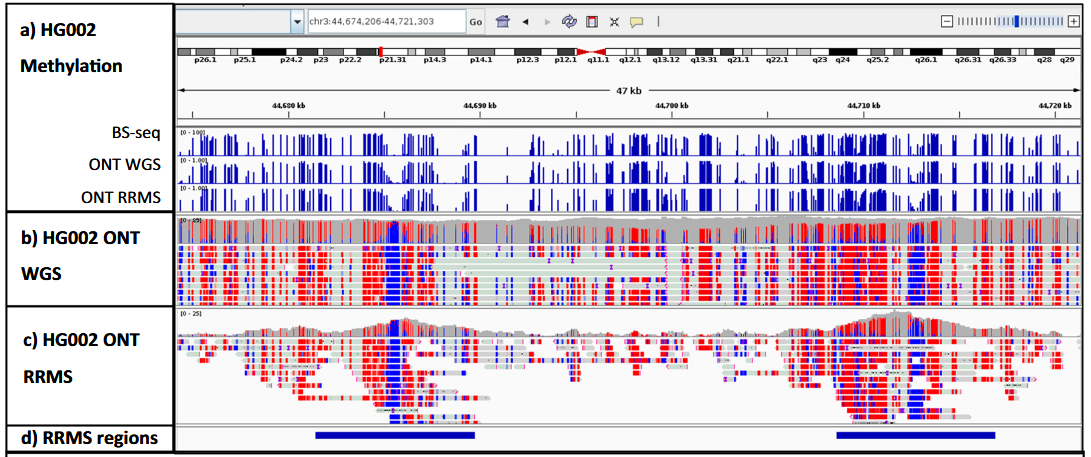
The figure shows the IGV (Integrative Genomics Viewer) plots of reads and methylation calls for whole-genome and RRMS (Reduced Representation Methylation Sequencing) nanopore sequencing, and compares the coverage of RRMS reads. Using DeepMod2, the authors detected 5mC in HG002 RRMS and found that 39 cytosines were detected within the whole-genome CpG motifs and 1399 were detected within the target regions. After aggregation, 248 CpG sites were detected across the whole genome and 717 were detected within the target regions. The number of aggregated CpG sites detected in the RRMS regions is more than that of stranded CpG sites, and the number of the latter is less than twice that of the former.
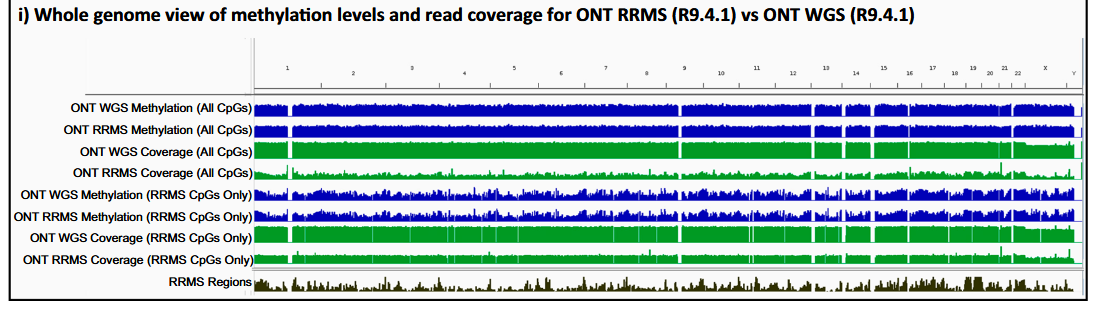
The figure displays the coverage and methylation levels of the ONT RRMS (Oxford Nanopore Technologies Reduced Representation Methylation Sequencing) and WGS (Whole Genome Sequencing) datasets, as well as the tracks of RMMS CpGs (Reduced Representation Methylation Sequencing Cytosine-Phosphate-Guanine sites) and all CpGs and the tracks of the RRMS target regions. The enrichment of the RRMS target regions relative to the off-target regions can be observed, as indicated by the coincidence of the RRMS tracks and the coverage peaks.