
RXBio Translates Sequence to Science and Industry

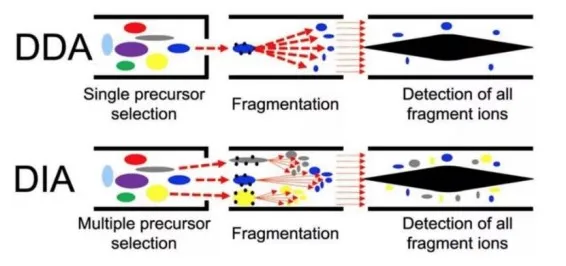
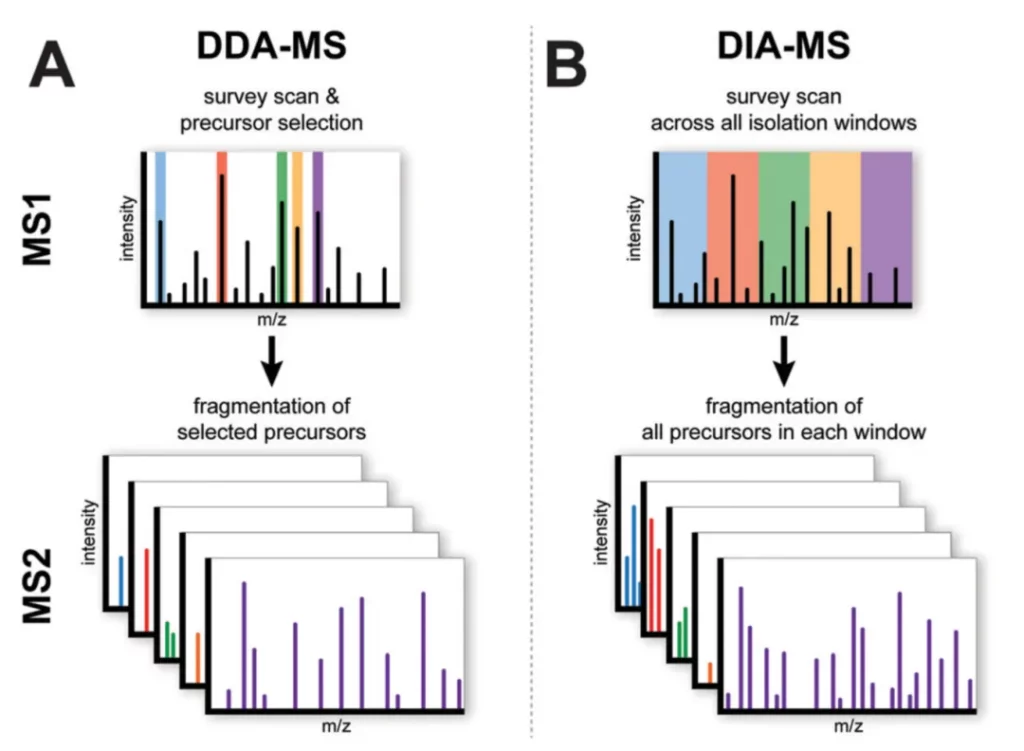
Data Independent Acquisition (DIA) is a brand-new mass spectrometry acquisition and quantification technique developed in recent years. Compared with the traditional Data Dependent Acquisition (DDA), it can overcome the bias towards the acquisition and fragmentation of high-abundance peptides and achieve the exhaustive acquisition of all peptide ions in samples, thus greatly improving the utilization of data and the accuracy of quantification.
✔ The DIA technique is not limited by the number of samples and can be used for the detection of a large number of samples.
✔ There are fewer sample pretreatment operations before sample preparation, which keeps the samples closest to their original states and improves the accuracy of detection.
✔ It enables panoramic scanning without missing any data, and greatly improves the accuracy of quantification.
✔ It has higher reproducibility, and the data can be traced back, which is beneficial to subsequent data analysis and mining.
✔ It will not lead to the loss of low-abundance proteins, and there are fewer missing values in the data.
The DIA (Data Independent Acquisition) technique utilizes high-resolution mass spectrometers to divide the entire full-scan range of the mass spectrum into several variable windows according to the distribution density of the mass-to-charge ratio (m/z). In each window, all peptide precursor ions will undergo ultra-high-speed, cyclic fragmentation and detection, thereby maximizing the scanning and collection of fragment information of all peptide ions. This panoramic scanning mode enables the DIA technique to obtain all the fragment information of all ions in the sample without omission, including that of low-abundance peptide ions.
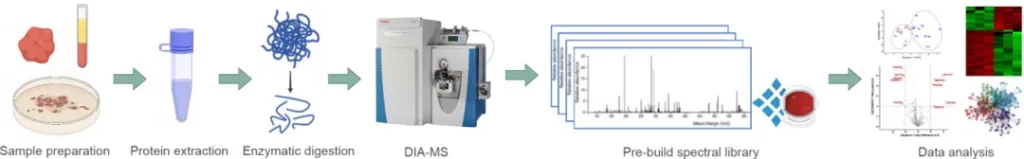
Protein extraction → Protein digestion → Mass spectrometry detection (in DIA mode) → Database search → Data analysis
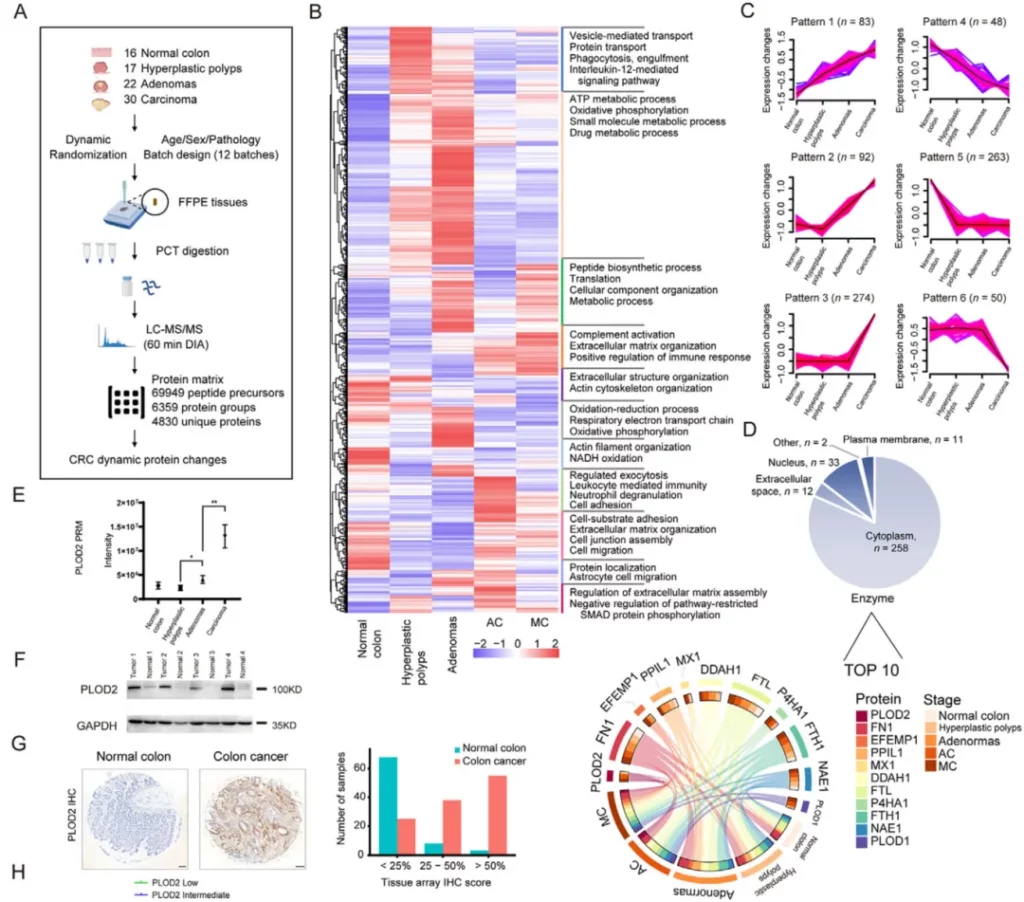
In February 2022, researchers from Westlake University, the School of Medicine of Zhejiang University and other institutions published an article titled “Proteomics profiling of colorectal cancer progression identifies PLOD2 as a potential therapeutic target” in Cancer Communications. In this study, the pressure cycling technology (PCT) was combined with the data-independent acquisition mass spectrometry (DIA-MS) technology to conduct proteomics analysis on colorectal cancer tumor tissue samples and characterize the proteomic dynamics related to the occurrence and progression of colorectal cancer.
The researchers compared the protein expressions in colorectal cancer samples at different clinical stages and those in normal colon tissue samples, and identified 928 differentially expressed proteins. They also demonstrated that these dysregulated proteins were mainly related to oxidative phosphorylation. Through unsupervised clustering analysis, six biologically significant protein expression patterns related to the occurrence and development of colorectal cancer were determined. Next, the researchers continued to narrow their focus to the proteins that were continuously upregulated during the tumor progression stage. They found that PLOD2 was the most upregulated protein and was consistently significantly upregulated in precancerous and cancerous samples compared with benign samples. This was later confirmed by parallel reaction monitoring (PRM) targeted proteomics technology, Western blot experiments and immunohistochemical staining.
Finally, combined with in vitro cell experiments and in vivo animal models, it was shown that PLOD2 contributed to tumor growth, resisted cell necrosis and was closely related to the development of colorectal cancer. PLOD2 was also involved in protein synthesis, metabolism and mRNA translation. In conclusion, through proteomics analysis and subsequent functional verification, this study identified PLOD2 as a potential new therapeutic target for blocking the carcinogenic process of colorectal cancer.
In June 2022, the Matthias Mann team published a research article titled “Noninvasive proteomic biomarkers for alcohol-related liver disease” in Nature Medicine. In this study, by using DIA proteomics combined with machine learning algorithms, they identified biomarkers with diagnostic value in detecting liver fibrosis, mild inflammatory activity, and steatosis. These three groups of biomarkers can detect any liver injury and help predict whether patients are at risk of disease progression, showing great potential for clinical application.
The researchers first analyzed the plasma samples of patients with alcohol-related liver disease (ALD) and healthy controls as well as the matched liver biopsy samples based on the DIA technique. By integrating liver-plasma proteome data, it was shown that 420 proteins appeared simultaneously in the liver and plasma quantitative proteomes, and the relative abundances of some of these proteins were strongly correlated in the liver and plasma. In addition, three groups of biomarkers were identified by using machine learning algorithms to identify significant fibrosis (nine proteins), mild inflammatory activity (six proteins), and any degree of liver steatosis (six proteins). Finally, the performance of the model was verified and the prognostic ability was evaluated in an independent cohort. In conclusion, this study demonstrated that the commonly dysregulated proteins in the liver and plasma represent different aspects of chronic liver disease and provided potential protein targets with diagnostic, prognostic, and therapeutic value in ALD.
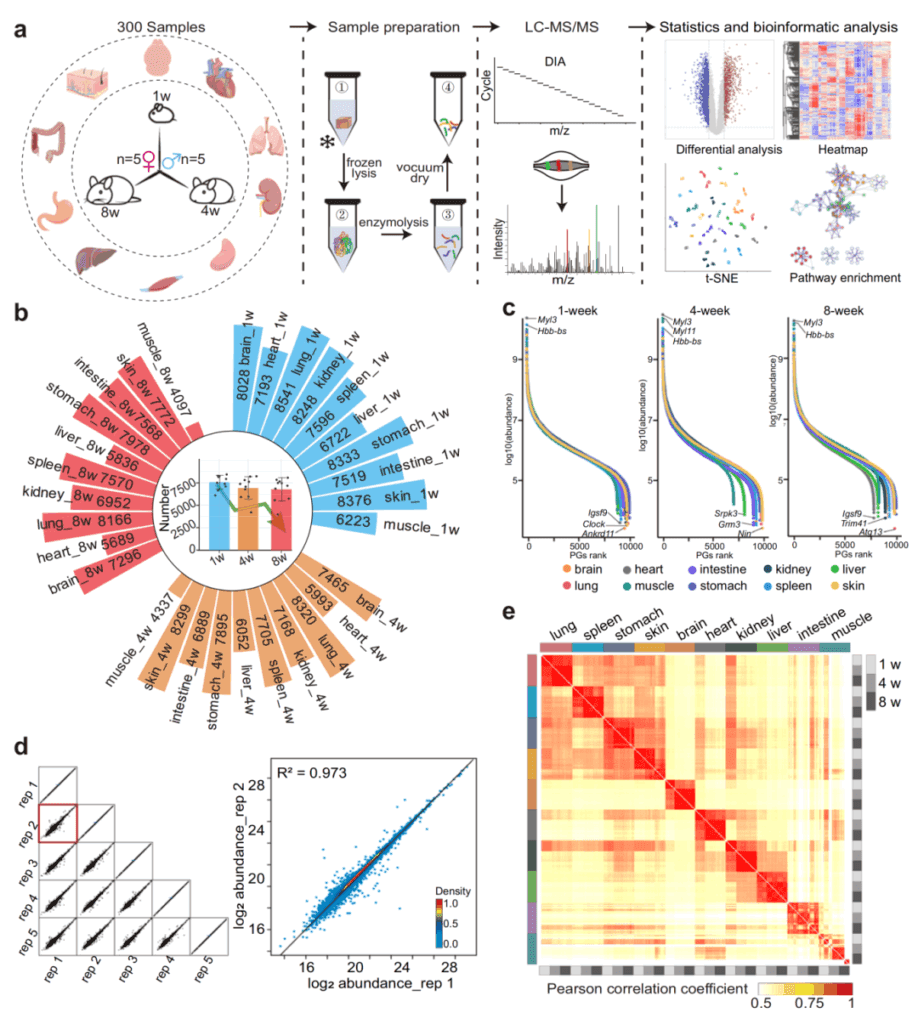
In July 2024, a team from the School of Biomedical Engineering at Shanghai Jiao Tong University published a research article titled “The mouse multi-organ proteome from infancy to adulthood” in Nature Communications. This study analyzed the proteomic changes of ten mouse organs at three key developmental stages through DIA proteomics. The research results provided a fundamental resource for understanding the molecular mechanisms of organ development and maturation in the early life.
The researchers generated a multi-organ proteome atlas of mouse development from infancy to adulthood based on the DIA technique. Through cross-comparison, 115 age-related differentially expressed proteins were detected in all organs, and it was shown that spliceosome proteins play a crucial regulatory role in the early development of multiple organs. In addition, protein expression patterns related to organ specificity and sexual dimorphism were also detected. These data revealed various identical and different biological processes involved in the growth and development of organisms, filling the research gap in the development and maturation of multiple organs in early life.
Reference
1. Shao, Y., et al. (2022). Proteomics profiling of colorectal cancer progression identifies PLOD2 as a potential therapeutic target. Cancer communications, 42(2): 164–169.
2. Niu, L., et al. (2022). Noninvasive proteomic biomarkers for alcohol-related liver disease. Nature medicine, 28(6): 1277–1287.
3. Wang, Q., et al. (2024). The mouse multi-organ proteome from infancy to adulthood. Nature communications, 15(1): 5752.
4. Krasny L, Huang PH. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol Omics. 2021;17(1):29-42.