恒河猴脑时空和性别相关lncRNA表达的注释及聚类分析_8-833x1024.png)
RXBio Translates Sequence to Science and Industry

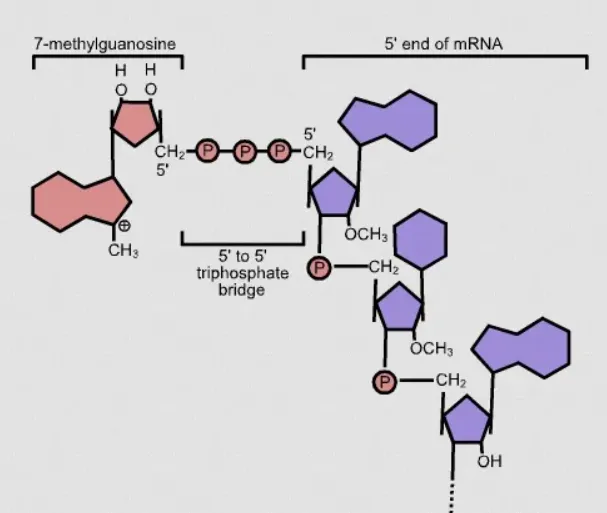
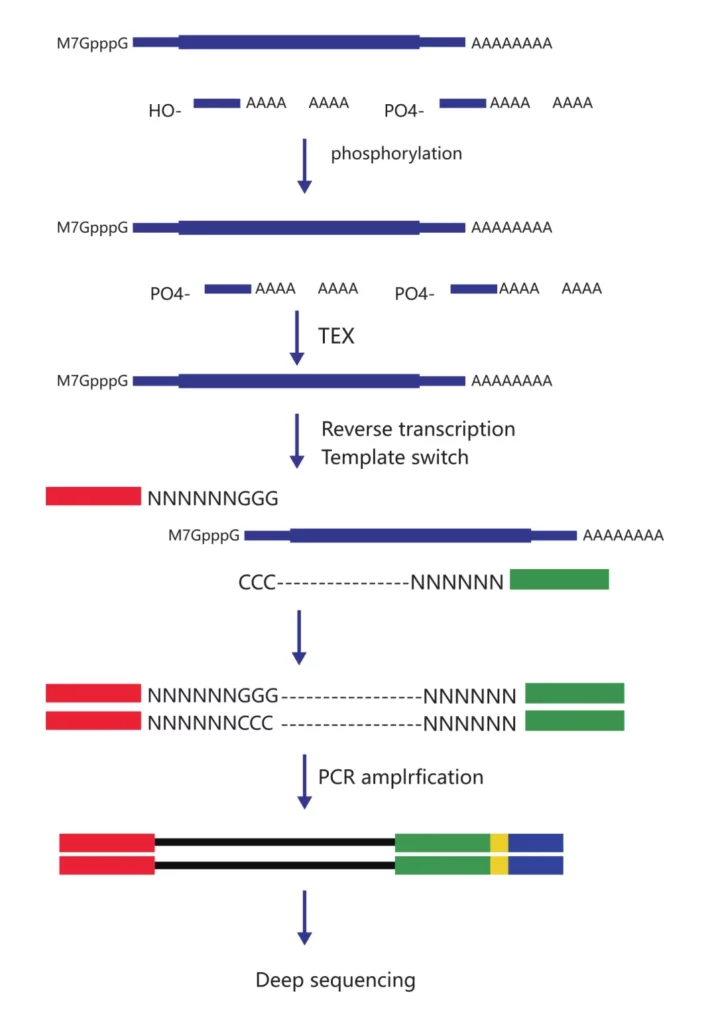
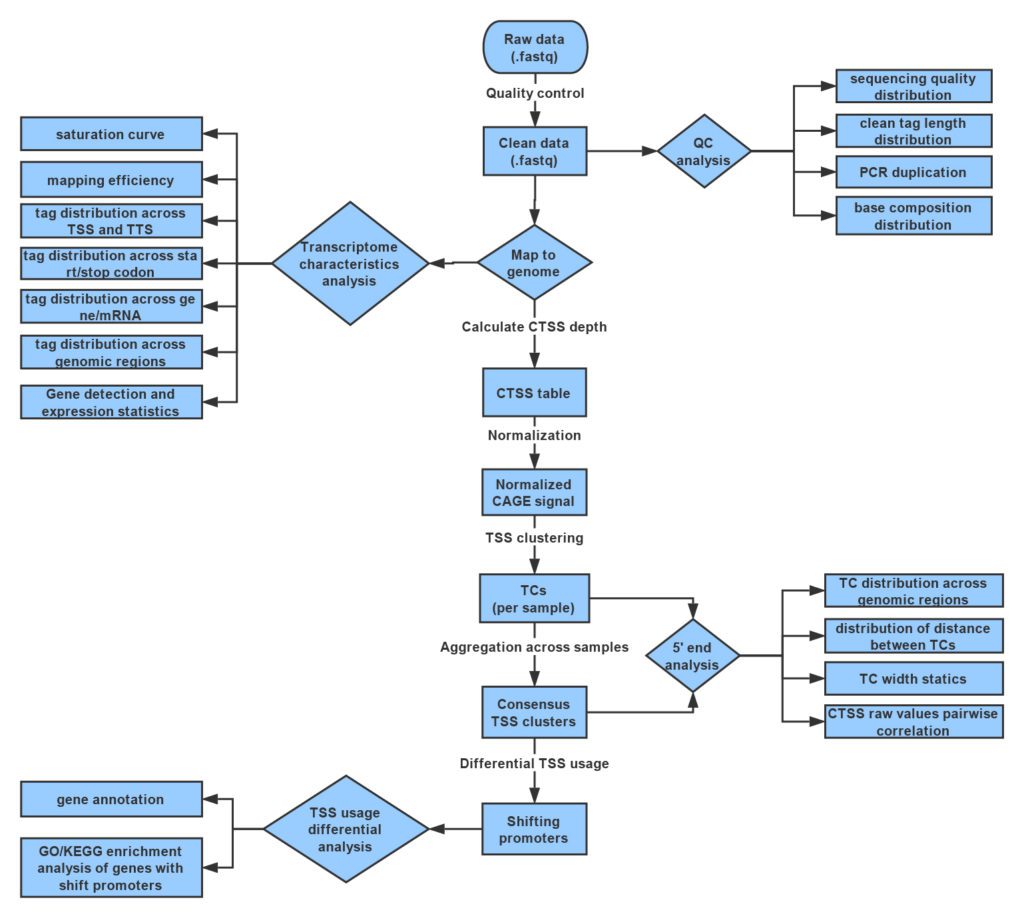
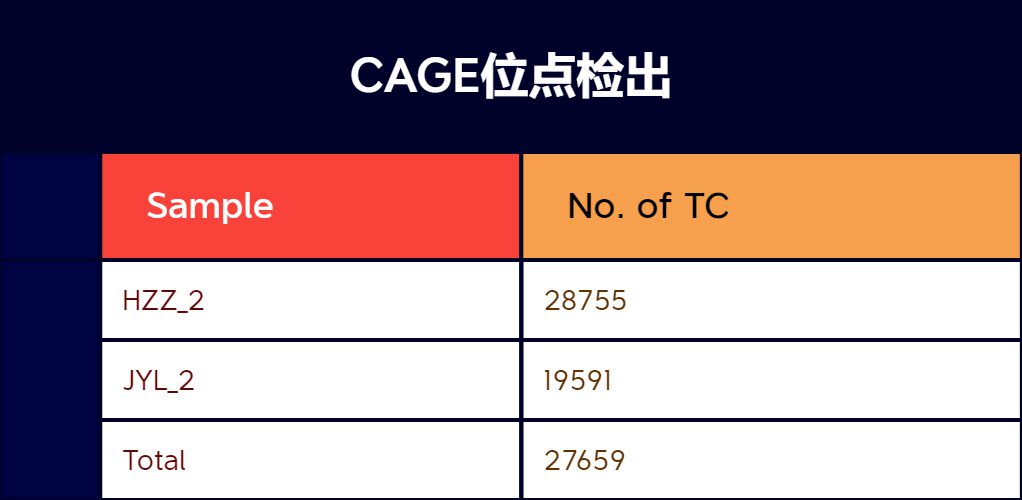
CAGE-seq,即加帽端mRNA测序,主要是鉴定转录起始位点(TSS)。绝大多数基因有两个甚至两个以上的转录起始位点,不同的转录起始位点会导致基因受到不同的上游非翻译区的调控作用(5’UTR)。不同的5’UTR序列中可能包含截然不同的作用元件,不同的起始位点导致了基因的表达所响应的信号也完全不同。同一个基因有可能受不同的启动子调控而导致表达的差异,可能会导致某些疾病的发生。CAGE-seq可以对mRNA中所有的TSS进行鉴定,发现新的启动子,以及差异表达分析,还可以推测可能的转录因子结合位点以及基因表达的网络调控。这是通过加帽位点鉴定实现的。
✔ 通过靶向转录起始位点(TSS)而不是整个基因,提供高度准确和详细的基因表达分析。据估计,人类基因组中大约有50,000个基因,但已确定的TSS超过185,000个。CAGE-seq 能够恢复大部分TSS。
✔ 基于每个TSS而不是每个基因的定量分析,可以发现microarray和RNA-seq无法检测到的差异表达基因。
✔ 不需要装载探针的DNA芯片,就可以对新基因进行分析。
✔ 提供了更广泛的动态范围来分析高表达和低表达的基因。
✔ 能够检测通常双向低水平表达的增强子RNA (eRNA)。
✔ TSS的精确鉴定使转录因子结合基序的预测比microarray更好。
2017年7月7日,《Genome Research》杂志在线发表了中国科学院昆明动物研究所李家立、胡新天和郑永唐学科组与中国科学技术大学生命科学学院汪香婷课题组合作的一篇研究论文,研究揭示了长链非编码RNAs在灵长类大脑发育和老化过程中表达动态变化和作用。
文章题目:Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expressiona in Rhesus macaque brain
中文题目:长链非编码RNAs在灵长类大脑发育和老化过程中表达动态变化和作用
期刊名:Genome Research(Q1 IF 11.9)
发表时间:2017年7月
实验材料:中国猕猴
研究策略
利用中国猕猴作为对象,通过RNA-seq和 CAGE-seq深度测序及分析,同时结合原位杂交与原代神经元系统的功能验证等一系列实验方法和手段,成功地解析了在中国猕猴大脑发育和老化过程中的动态变化特征。
研究发现
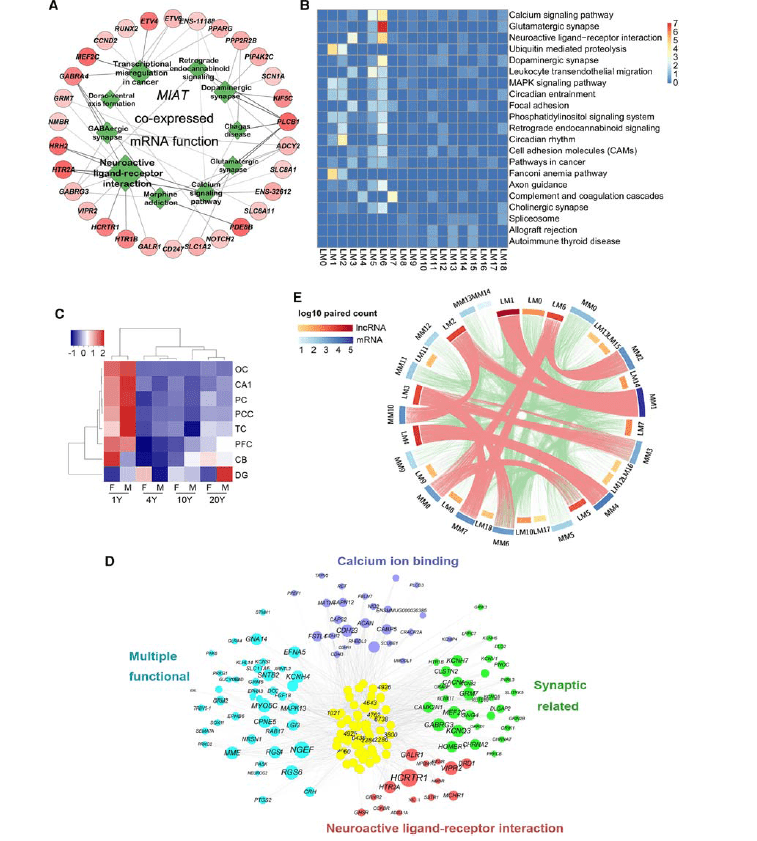
大脑特异性lncRNAs呈现出高度的区域、性别和年龄表达差异性。
依据这些表达差异性,总共发现18个不同的 lncRNAs和14个不同的mRNAs表达模块式。相对于mRNAs的表达变化特征,猕猴脑特异性lncRNAs的表达性状在发育和老化过程中呈现出更高的动态变化特征。
在此基础上,利用CAGE-seq对lncRNA和mRNA启动子区域进行测序和分析,发现lncRNAs的启动子区域变化比mRNA呈现出更明显的性别差异和时空变化性状。
进一步研究还发现,lncRNAs和 mRNAs之间存在多重的正相关和负相关调控关系,其中在大脑皮层区域特异性表达的lncRNAs和 mRNAs之间负相关的调控关系对发育和老化过程中大脑皮层的结构和神经元功能变化具有重要的调控作用。
研究意义
上述研究结果为进一步认识和理解灵长类动物复杂的大脑结构和高级认知功能的表观调控基础提供了新的实验依据,也为进一步探索与脑发育、和衰老相关的神经系统疾病的发病机理和治疗新靶点提供了新的研究方向。

电话:027-870502099
邮箱:sales@rxbio.cc
地址:武汉市东湖高新区高新二路388 号
光谷生物医药加速器 18 栋 1-2层
单细胞多组学 空间转录组
三代测序 功能基因组
表观遗传学 互作组学
单细胞大数据 数据深度挖掘

欢迎关注公众号「瑞兴生物」