
RXBio Translates Sequence to Science and Industry

真核细胞中,大多数蛋白质编码基因和长链非编码RNA的3’端会经过切割和聚腺苷化(polyA),最近的研究表明绝大多数的mRNA具有两个或以上的聚腺苷化位点(PAS),因此选择性多聚腺苷酸化( APA,是指具有多个PAS的基因),是真核生物的一个重要转录后调控方式。它广泛存在于所有真核生物中,是基因调控的主要机制。
APA事件不仅可导致编码基因产生出多个3’UTR长度和序列组成不同的转录异构体,拓展蛋白质组学和功能多样性,而且在基因调控中起着重要的作用。作为一种关键的分子机制,APA参与了mRNA成熟、mRNA稳定、细胞RNA衰变和蛋白质多样化等多种基因调控步骤。APA在癌症中经常出现失调,导致癌基因和肿瘤抑制基因表达的改变;正常细胞中癌基因通常使用远端PAS形成长3’UTR亚型,而肿瘤细胞往往使用近端PAS而形成短3’UTR亚型。3’UTR的缩短可能显著减少了miRNA的抑制作用,使mRNA更趋于稳定,翻译效率显著提高,使相关基因蛋白质表达水平升高。
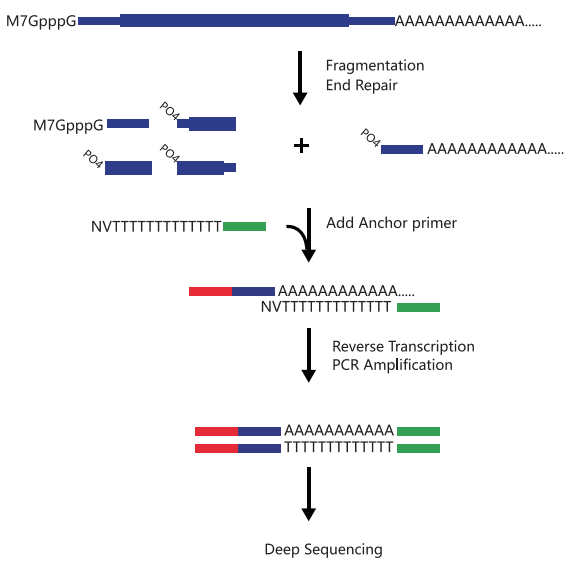
PAS-seq,是一种检测可变的多聚腺甘酸位点的技术方法。可变的多聚腺甘酸化(alter-native polyadenylation,APA)是真核生物中一种重要的基因表达调控方式。通过PAS-seq以及后续的信息学分析,我们可以得到转录组范围内的转录终止位点(TTS)、不同的3’UTR分析、差异表达分析。
✔ 直接、便捷
✔ 容易实现
✔ 自有分析流程
✔ 个性化服务支持
选择性多聚腺苷化(APA)在抑制宫颈癌中的重要调控作用
Journal:Oncogene,IF=9.867
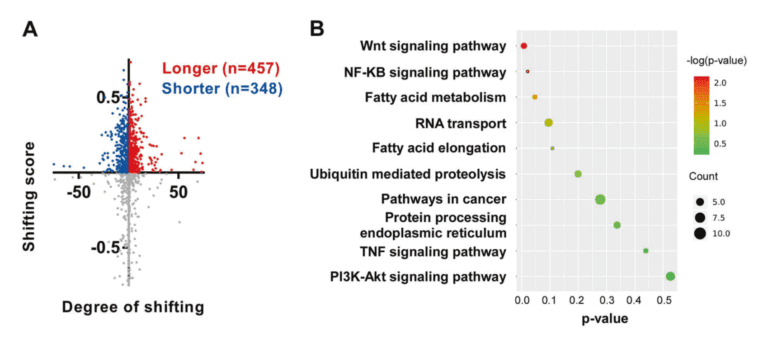
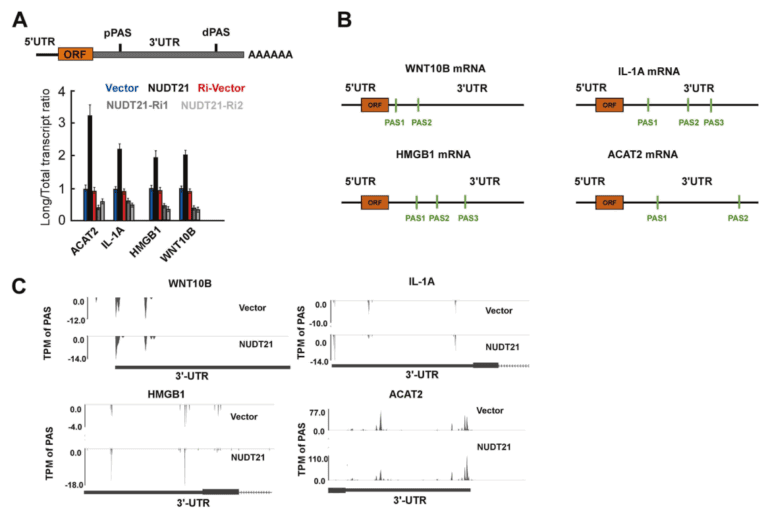
运用我公司特有的PAS-seq技术在宫颈癌全基因组范围内对聚腺苷化RNA的3个末端区域(3’UTR)进行测序,绘制响应NUDT21过表达和敲低的APA事件,随后进行功能注释分析。在NUDT21过表达情况下,发现457个转录本的3’UTRS显著延长。与此同时,348个转录本的3’UTRS显著缩短。通过功能注释分析发现NUDT21靶点参与了脂肪酸(FA)代谢调控因子和Wnt和NF-KB信号通路的作用。从以上的APA受NUDT21调控的基因中,作者进一步确认IL-1A、WNT10B、HMGB1和ACAT2参与宫颈癌转移基因。NUDT21可调控CxCa细胞中miRNA介导的翻译抑制。

电话:027-870502099
邮箱:sales@rxbio.cc
地址:武汉市东湖高新区高新二路388 号
光谷生物医药加速器 18 栋 1-2层
单细胞多组学 空间转录组
三代测序 功能基因组
表观遗传学 互作组学
单细胞大数据 数据深度挖掘

欢迎关注公众号「瑞兴生物」