
看得懂字,却看不懂“话”?
本文将带你系统掌握高通量测序(NGS)中的关键入门术语~
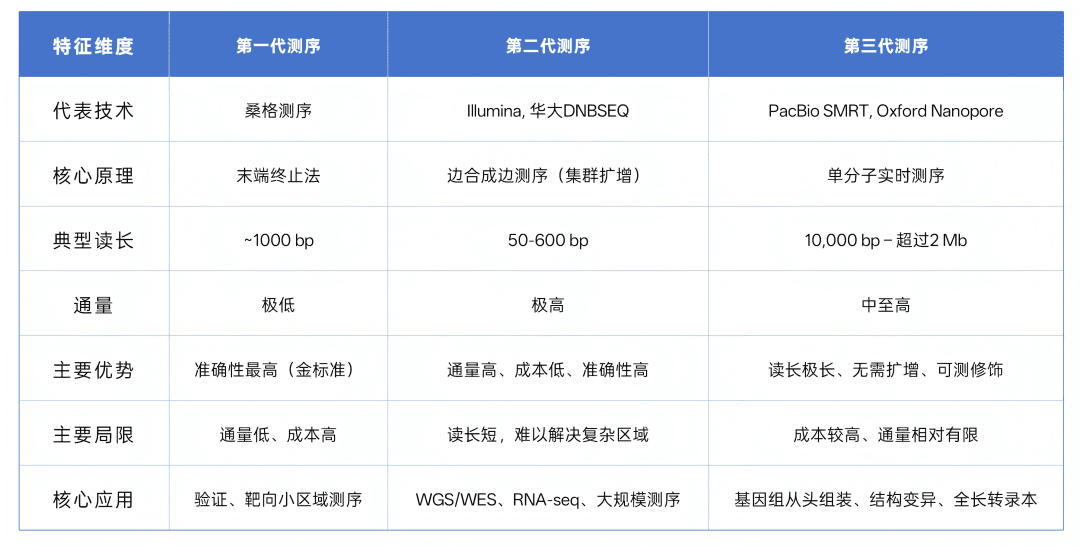
测序技术发展
从 Sanger 到 NGS 再到长读长
第二代测序(NGS)以Illumina和华大基因为代表,通过边合成边测序技术实现大规模并行测序,将通量提升数个数量级的同时大幅降低成本,成为当今基因组学研究与临床检测的绝对主流。其核心优势在于高通量、高准确性,但读长较短。
第三代测序以PacBio和Oxford Nanopore为代表,实现了单分子实时测序,无需PCR扩增。其革命性优势在于超长读长,能够跨越复杂基因组区域,直接检测表观修饰,在基因组完整组装、结构变异解析等领域具有不可替代的价值。
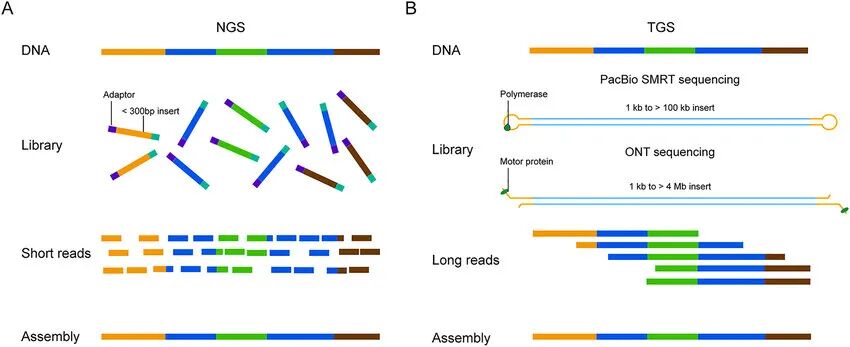
测序数据基础
什么是 Reads?PE?
● 二代短读长平台:通常为50~300 bp,适合高精度、高通量的项目。
● 三代长读长平台:可长达 10 kb 以上 ,适合结构变异、全长转录本分析等。
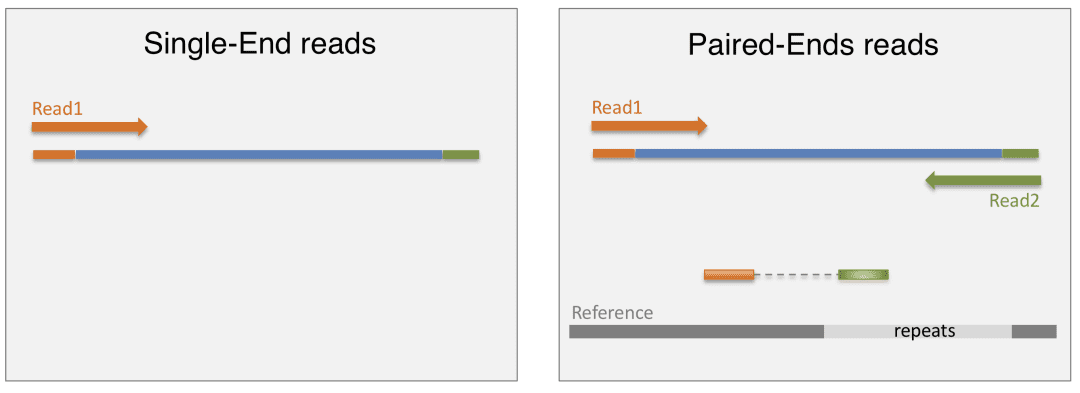
● SE:仅测一端,成本低,适用于小RNA、miRNA等短序列项目。
选择建议:若预算允许,优先选择PE测序,数据利用率与可靠性更高。
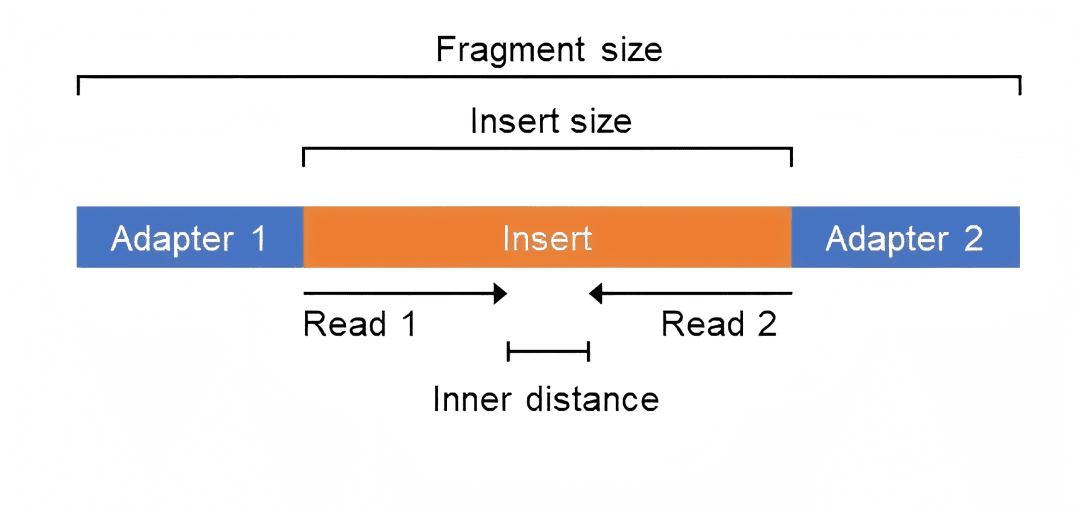
样本 → 文库 → 上机
建库是数据质量的根本
● DNA建库流程(以全基因组测序为例)
DNA样本 → 片段化 → 末端修复 & 3’端加”A” → 连接接头 → PCR富集 → 文库质控 → 上机测序
● RNA建库流程(以mRNA转录组测序为例)
Total RNA → mRNA富集(或rRNA去除)→ 片段化 → 反转录为cDNA → 双链cDNA合成 → 末端修复 & 3’端加”A” → 连接接头 → PCR富集 → 文库质控 → 上机测序
● WES全基因组测序:300–500 bp
● RNA-seq转录组测序:200–300 bp
合适的插入片段能提升测序效率与数据均匀性。
● 单端Index:一个样本一个标签
● 双端Index:两端各一个标签,识别更精准
Index技术是实现“多样本混测”的核心,极大降低了单位样本成本。
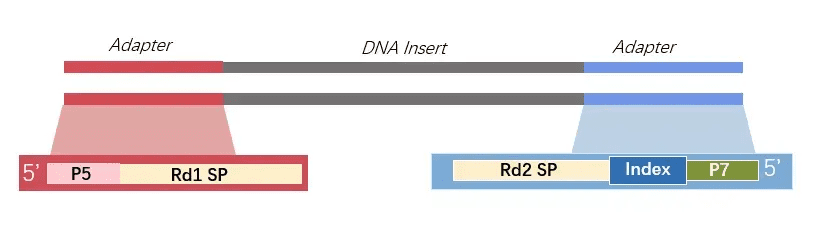
单端index文库结构
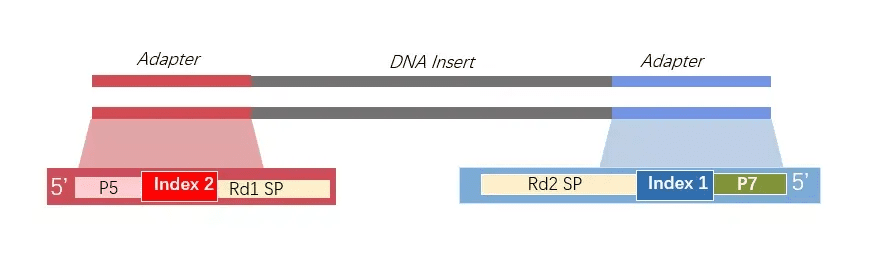
双端index文库结构
数据分析的核心指标
● 30X 覆盖度:表示平均每个碱基被测序30次。
● 高覆盖度有助于发现低频突变,提高检测灵敏度。
● 肿瘤基因突变检测
● 稀有变异挖掘
● Q30:错误率为1/1000, 99.9% 准确
● Q20:错误率为1/100, 99% 准确
常以“≥85% Reads 达到 Q30”作为高质量数据标准。
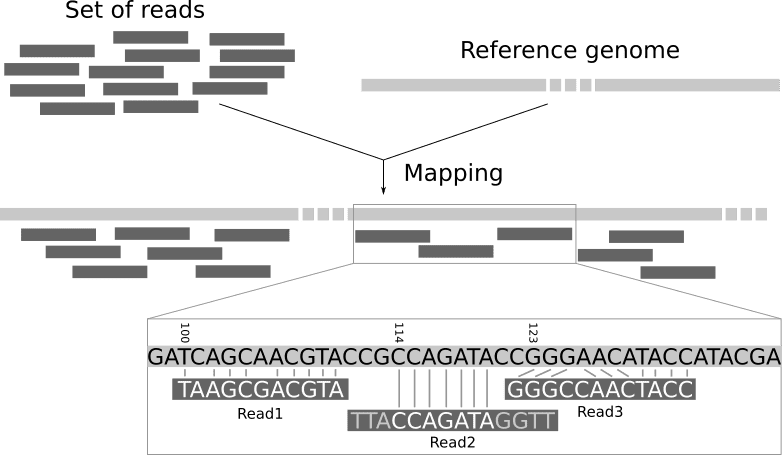
比对率:成功比对到参考基因组的Reads比例。低的比对率可能提示样本污染或参考基因组选择不当。
基因组变异
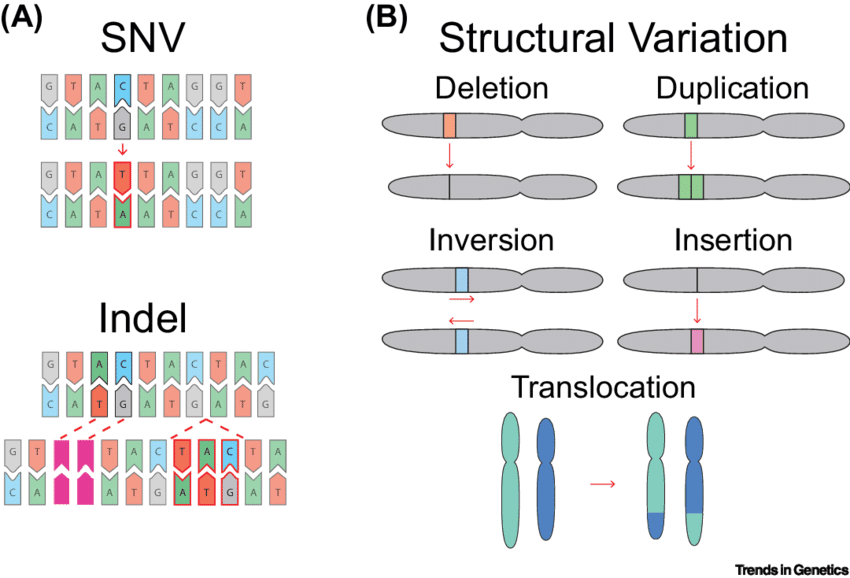
读懂 SNP、SNV、InDel、CNV、SV
● SNP:特指那些在群体中发生频率大于1% 的SNV。通常指代在人群里常见的、遗传下来的多态性位点。
简单理解:所有的SNP都是SNV,但并非所有的SNV都是SNP。SNP更强调“群体普遍性”,而SNV是一个更中性的技术术语。在肿瘤研究中,体细胞突变常称为SNV;在群体遗传中,则常用SNP。
● 关键影响:在编码区的InDel如果不是3的倍数,会导致移码突变,从而可能造成蛋白质功能的严重破坏。
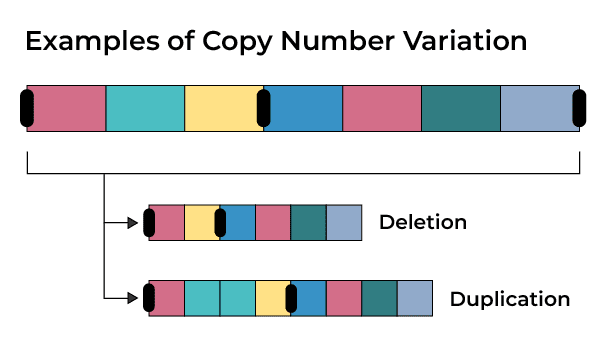
● CNV:指基因组上一段DNA序列(通常>1kb)的拷贝数目相较于参考基因组发生了变化,包括缺失和重复/扩增。
○ 应用:是癌症基因组(如致癌基因的扩增)和遗传病研究中的重要标志物。
● SV:指基因组发生的大尺度结构改变,通常涉及>1kb的DNA片段。
○ 主要类型:
ⅰ. 缺失 / 重复(与CNV有重叠)
ⅱ. 倒位:一段序列被反向插入。
ⅲ. 易位:两段来自不同染色体或同一染色体不同位置的序列发生交换。
○ 应用:是许多遗传性疾病和癌症的重要驱动因素。
简单理解:CNV主要关心“数量” (一段DNA有几份拷贝);SV主要关心“结构”与“位置” (DNA片段是否被重排了)。易位和倒位属于SV,但不属于CNV;大的缺失/重复则既是SV也是CNV。
转录组重要概念
转录本与基因
● 过程:DNA(基因)→ 通过RNA聚合酶转录 → 初级RNA → 经过加工 → 成熟的RNA(即转录本)。
● 主要类型:对于编码蛋白质的基因,其最主要的转录本就是信使RNA。因此,在转录组测序中,我们通常所说的“转录本”即指成熟的mRNA。
主要原因和机制如下:
1. 可变剪接(核心机制):
○ 基因由外显子和内含子构成。转录后,初级RNA需要剪接,即“切除”内含子,“拼接”外显子。
○ 可变剪接允许细胞在不同的发育阶段、组织类型或环境信号下,选择性地将不同的外显子组合在一起,从而从一个基因产生多个外显子组成不同的mRNA。
2. 可变转录起始位点:
○ 基因可能有多个“启动开关”。选择不同的转录起始位点,会生成具有不同第一个外显子的mRNA。
3. 可变多聚腺苷酸化位点:
○ mRNA的3‘端需要添加一串“A”(多聚腺苷酸尾)。选择不同的加尾位点,会生成具有不同长度3’端非翻译区甚至不同末端外显子的mRNA。
● 精细调控:有些转录本可能包含影响mRNA稳定性、翻译效率或亚细胞定位的序列,实现对基因表达的精细调控。
● 适应复杂生命活动:这是高等生物用有限的基因数量(人类约2万个)产生极其复杂的蛋白质组和生命现象的核心策略。
表观组和转录因子研究
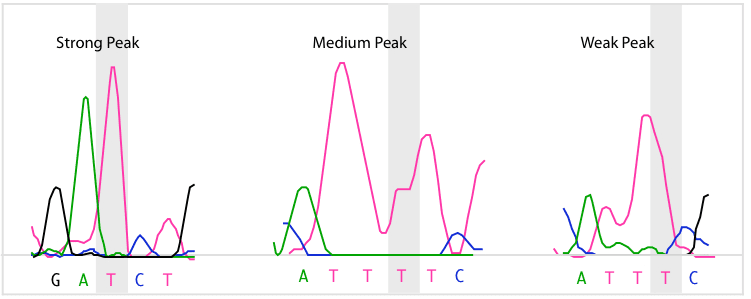
怎么看信号图
1. 坐标轴:
○ X轴:基因组位置,通常会标注基因、启动子等关键区域。
○ Y轴:测序读数的富集强度(或信号值)。
2. 图中的“山峰”:
○ 峰的存在:有“山峰”的区域就是潜在的蛋白质结合或开放染色质区域。
○ 峰的高度:峰越高(Y轴值越大),代表该位点的结合强度越强或染色质可及性越高,通常也意味着该位点越重要。
○ 峰的宽度:峰越宽,代表结合的基因组区域越宽。
3. 对比样本:
○ 实验组:通常是有色的轨迹(如红色、蓝色),你会在目标区域看到明显的“山峰”。
○ Input/对照组:通常是灰色或黑色的轨迹,它代表背景信号,应该是平坦或无规则的。只有当实验组的峰显著高于Input对照组时,这个峰才是可信的。
一句话解读:寻找那些在实验组中高高耸起、而在对照组中平坦如初的“山峰”,它们就是你的目标功能区域。
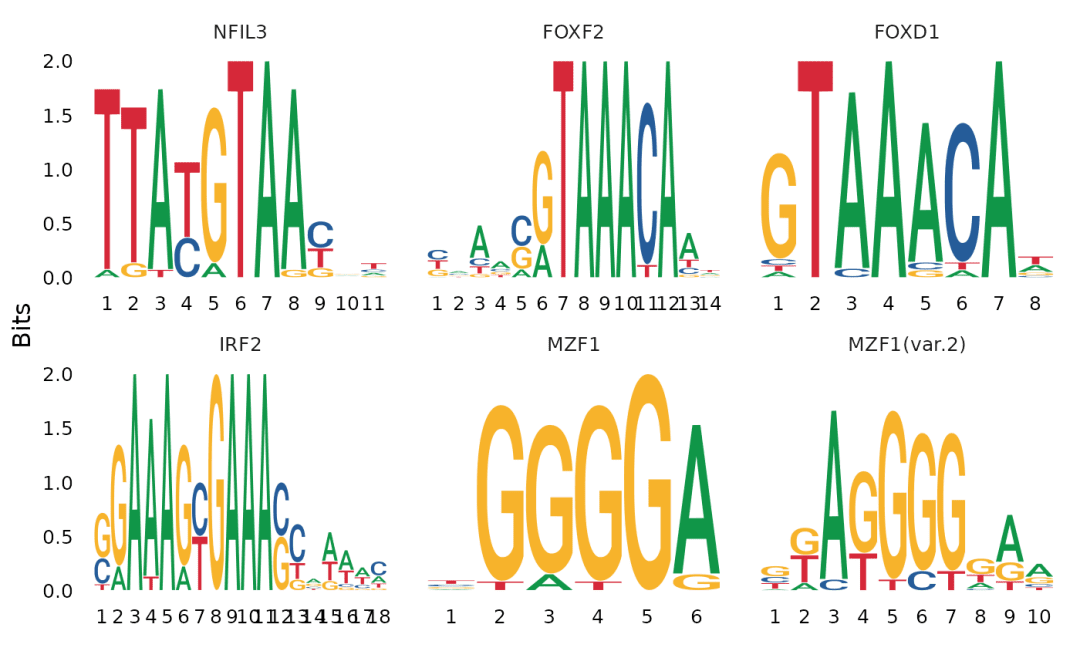
1. 每个字母的“高度”:
○ 在序列的每个位置上,A, T, C, G四个字母堆叠起来的总高度代表该位点的保守程度或信息量。总高度越高,说明这个位置越重要,碱基选择“挑剔”。
2. 单个字母的“大小”:
○ 在每个位置上,字母本身的大小与该碱基在此位置出现的频率成正比。字母越大,说明这个碱基在此处越常见。
○ 例如: 如果在某个位置只有一个巨大的“A”,其他三个字母很小,说明几乎所有序列在这个位置都是A,此处高度保守。
3. 整体模式:
○ 观察整个序列模式,它代表了结合蛋白偏好的“序列指纹”。
○ 例如,一个锌指蛋白的Motif可能呈现特定的模式,而一个碱性亮氨酸拉链蛋白的Motif则可能是另一种模式。
一句话解读:看哪个碱基在每个位置上“最大”、最突出,整个堆叠起来的高序列模式就是推测出的转录因子“结合密码”。
数据格式
FASTQ、SAM、BAM
● SAM为文本格式,可读性强;
● BAM为二进制格式,压缩存储,便于传输与分析。
![]() 若有所获,欢迎点赞、推荐与分享
若有所获,欢迎点赞、推荐与分享