
RXBio Translates Sequence to Science and Industry

通过对⾎液、组织或细胞样本进行ONT全基因组重测序,不仅可以分析CNV、SV、SNP和结构变异等,还可以在单碱基水平检测DNA各类修饰。
全基因组重测序(Resequencing) 是对具备已知参考基因组序列的物种个体进行基因组测序,通过与参考序列比对,分析个体或群体间的基因组差异。该技术能够高效、全面地检测基因组中的多种变异类型,包括:单核苷酸多态性(SNP)、拷贝数变异(CNV)、插入/缺失(InDel)、结构变异(SV)。凭借其强大的变异检测能力,全基因组重测序已广泛应用于临床医药研究、群体遗传学、关联分析、进化分析等多个领域。
二代测序(NGS)短读长(≤300 bp)难以解析重复序列和复杂结构变异,PCR扩增偏差影响拷贝数变异(CNV)等检测灵敏度。三代测序(LRS)凭借超长读长精准解决复杂区域,无PCR偏差提升覆盖均匀性,且高准确性(>99%)支持临床级应用,在结构变异、单倍型分型等方面优势显著。
ONT全基因组重测序(ONT-lrWGRS)是利用牛津纳米孔技术(ONT)对基因组DNA进行直接测序的方法。其核心优势在于超长读长,能有效跨越复杂重复区域和解析结构变异,同时可检测碱基修饰(如甲基化),适用于基因组组装、结构变异鉴定和表观遗传学研究,具有实时、快速的特点。
✔ 高变异检出率:无需PCR扩增,无GC偏好,超长读长,轻松跨过高重复和高复杂度区域。
✔ 精确的变异信息:超长读长,为检测检测SV大结构变异提供保障。
✔ 更丰富的分析内容:除变异信息之外,还能检测DNA上的各种碱基修饰。
1. 文库构建原理
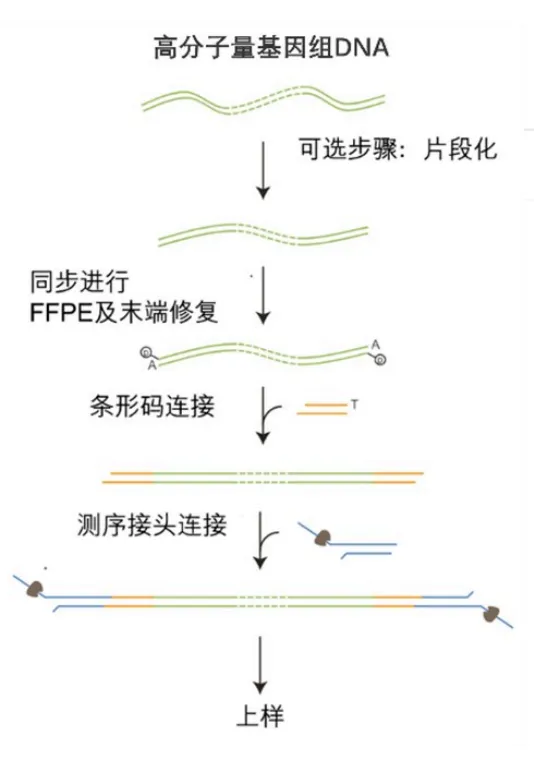
图1:ONT全基因组重测序文库构建流程图
首先提取高质量的基因组DNA样本,对DNA样本进行打断(可选)。接着对DNA样本进行末端修复和加A,连接样本Barcode,连接测序接头。加载到测序芯片上进行测序。
2. 技术路线
• 提取的DNA样本:总量≥5 ug,浓度≥200 ng/uL,纯度:260/280 ≥1.8、细胞样本:≥1×107cells
• 新鲜或冻存的动物组织样本:≥200 mg
• 新鲜或冻存的植物组织样本:≥500 mg
• 全血:≥ 4 mL
案例1
文献1: Structural variants in the Chinese population and their impact on phenotypes, diseases and population adaptation
中文标题:中国人群的结构变异及其对表型、疾病和群体适应的影响
期刊:Nature Communications
影响因子:14.7
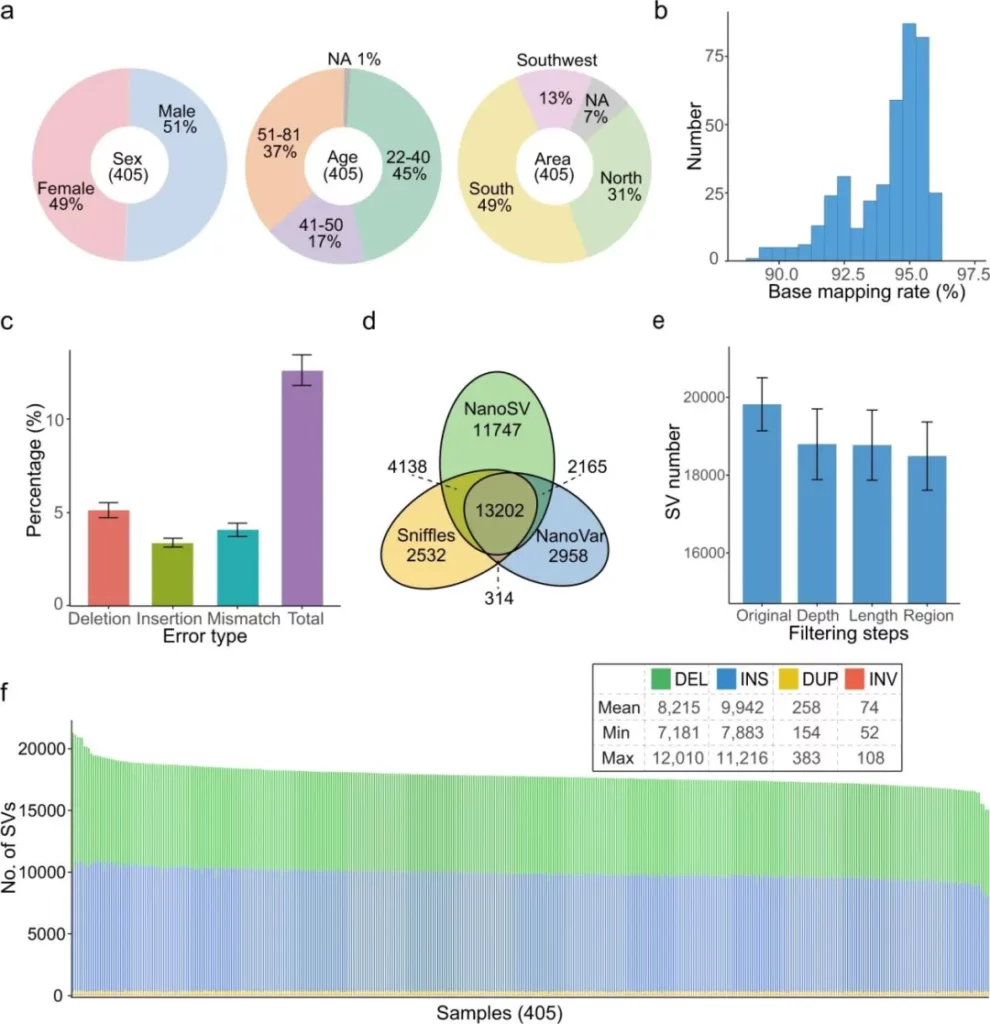
样本:本研究共纳入405人(男性206人,女性199人),年龄从22岁到81岁不等
测序平台:Oxford Nanopore PromethION
作者基于ONT平台开展全基因组长读长测序,结合健康筛查获取327个个体的68项表型与临床测量值,构建了包含基因组测序数据与表型/临床数据的综合数据库。数据经质控后,将干净读长比对至参考基因组GRCh38,检测出长度≥50bp的DEL(缺失)、INS(插入)、DUP(重复)和INV(倒位)四类结构变异(SV)。
为提升SV可靠性,采用Sniffles、NanoVar和NanoSV三种长读长测序分型方法交叉验证,仅保留被至少两种方法识别的SV,有效降低假阳性率。结果显示,平均每个样本鉴定出18,489个高置信度SV,数量近似正态分布,以DEL和INS为主。进一步分析发现,当测序深度超过15倍时,SV数量增长趋于饱和,表明深度与SV检出率间存在非线性关联。研究结果为基因结构变异特征分析提供了定量依据,并揭示了测序深度对SV检测效率的影响规律。
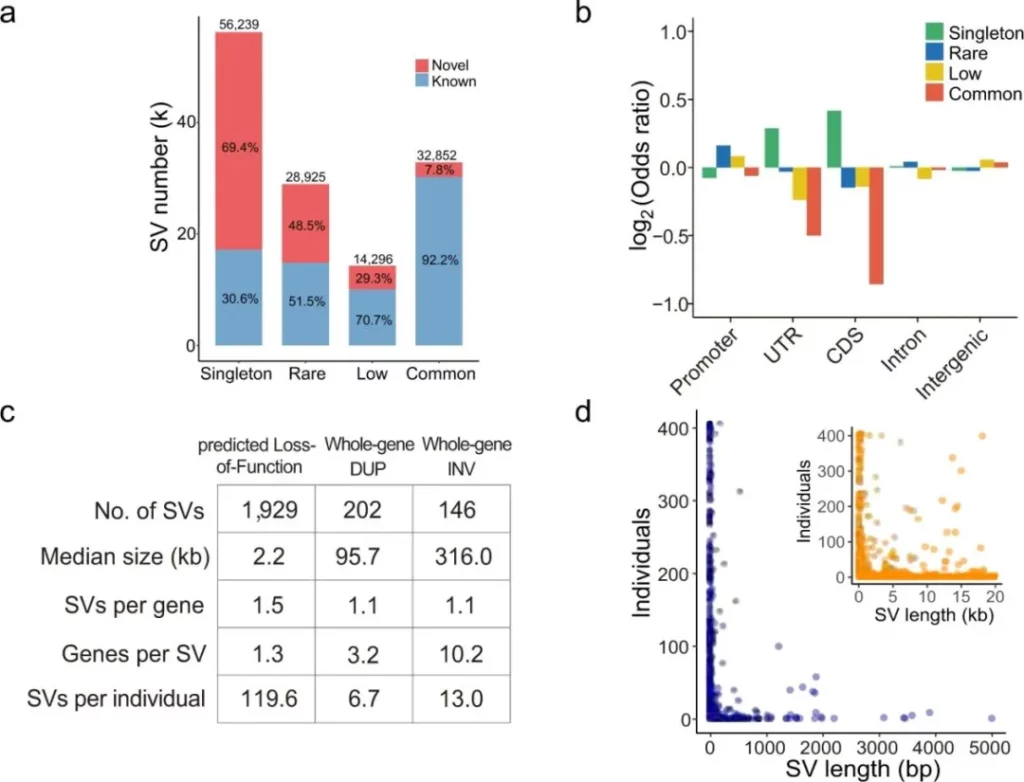
为解析结构变异(SV)的潜在功能,作者基于基因组位置对SV进行注释,覆盖编码区(CDS)、非翻译区(UTR)、启动子及内含子区域。结果显示,内含子区SV占比最高,而UTR和CDS区的SV中,单例SV(仅在单个样本中检测到)显著富集,提示其潜在功能倾向性。针对与CDS重叠的SV,依据断点位置进一步划分为三个亚型:预测功能丧失型(pLoF):导致编码核苷酸缺失或开放阅读框改变;全基因复制型(WDUP):引发基因整体拷贝数增加;全基因倒位型(WINV):通过重组上游增强子与基因位置/方向调控表达。功能注释发现,受pLoF SV影响的38个基因在“免疫球蛋白受体结合”通路显著富集,暗示其在免疫调控中的潜在作用。
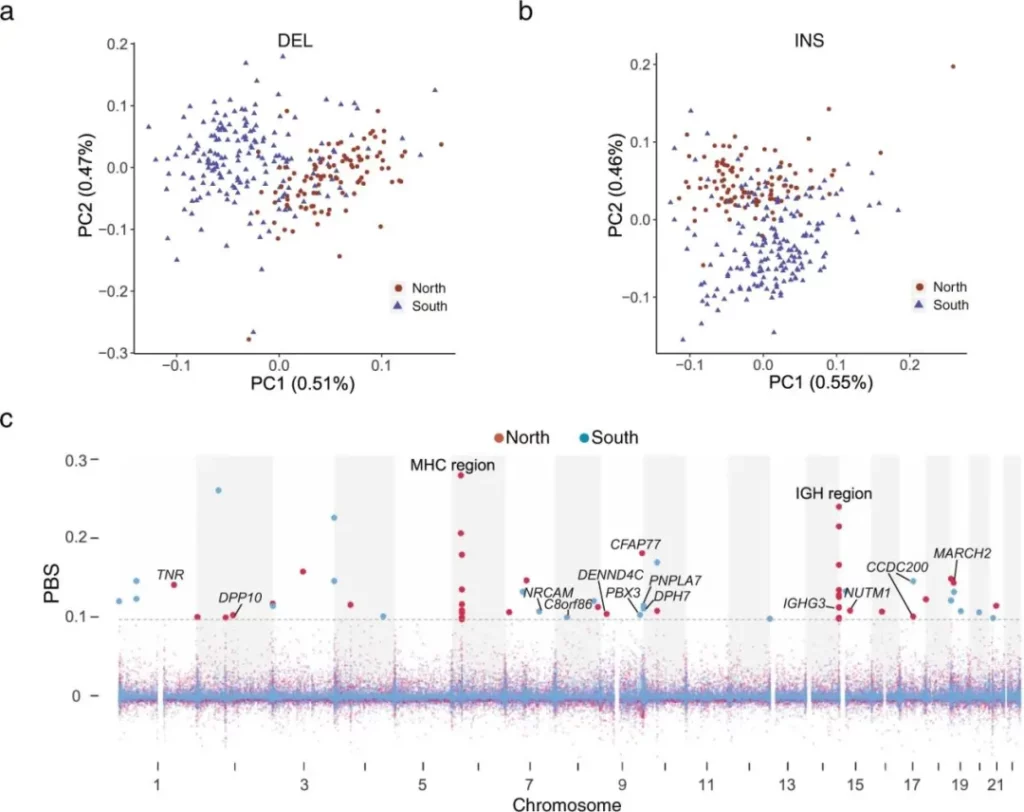
为了更好地理解pLoF SV如何影响临床表型和疾病,作者使用GWAS catalog,OMIM和COSMIC数据库注释了SV及其相关基因。例如,一个19.3 kb的杂合罕见DEL,覆盖了血红蛋白亚基α 1和2(HBA 1和HBA 2),其功能障碍会导致α-地中海贫血; 27.4kb的杂合DEL,其含有血红蛋白亚基β(HBB),其功能障碍会引起严重的血红蛋白病,例如镰状细胞贫血和β-地中海贫血。另外,在gnomAD 数据库中发现19.3kb的DEL序列,其中92.3%来自东亚群体,这表明该DEL序列具有群体特异性。最后进一步基于次要等位基因频率(MAF)> 0.05的基因分型SV进行了临床表型的GWAS,例如生化、血液和尿液组成,还探讨了中国北方和南方群体之间的群体遗传特性的基础上DEL和INS。PCA显示两组之间存在明显的遗传多样性;华南、华北和华南与华北之间个体的平均IBS距离较小,表明亚群之间的差异很小。
虽然之前使用SNP阵列和基于SRS的WGS已经揭示了中国北方和南方个体之间的遗传差异,但本文作者使用FST和PBS分别在中国北方和南方人群的基因组中观察到24个和35个独立的PBS信号。在中国北方人群中,前两个信号位于主要组织相容性复合体(MHC)区域(6p21.3-p22.1)和免疫球蛋白重链(IGH)簇基因座(14q32.33)。在MHC区域中,具有PBS信号的九个SV位于HLA-G、HLA-A、HLA-DRA、HLA-DRB 5、HLA-DRB 1、HLA-DQA 1和HLADPA 1的基因间区域。值得注意的是,有10个具有PBS信号的SV位于IGH簇基因座中,例如IGHG 3和TEDC 1、TMEM 121、IGHA 2、IGHE、IGHG 2、IGHA 1、IGHG 1和IGHG 3的基因间区,这表明IGH基因不同基因型的积累和组合可能与免疫适应不同环境有关。在中国南方人群中也检测到MHC和IGH区域的PBS信号。这一结果表明,免疫相关区域的SV可能是由于长期暴露于不同的环境而产生的。此外,具有1.4kb INS的PBS信号位于PNPLA 7的第13内含子中。先前基于RNA测序分析,PNPLA 7与高血压相关。而中国北方人口的血压和高血压患病率高于南方人口。但需要更多的证据来证实PNPLA 7内含子中的INS可以引起基因表达的变化,从而导致血压的变化。
案例2
文献2: Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease
中文标题:长读测序鉴定与神经元核内包涵病相关的NOTCH2NLC中GGC重复扩增
期刊:Nature Genetics
影响因子:31.7
样本:8个家庭的13个患者和4例正常个体
测序平台:Oxford Nanopore PromethION
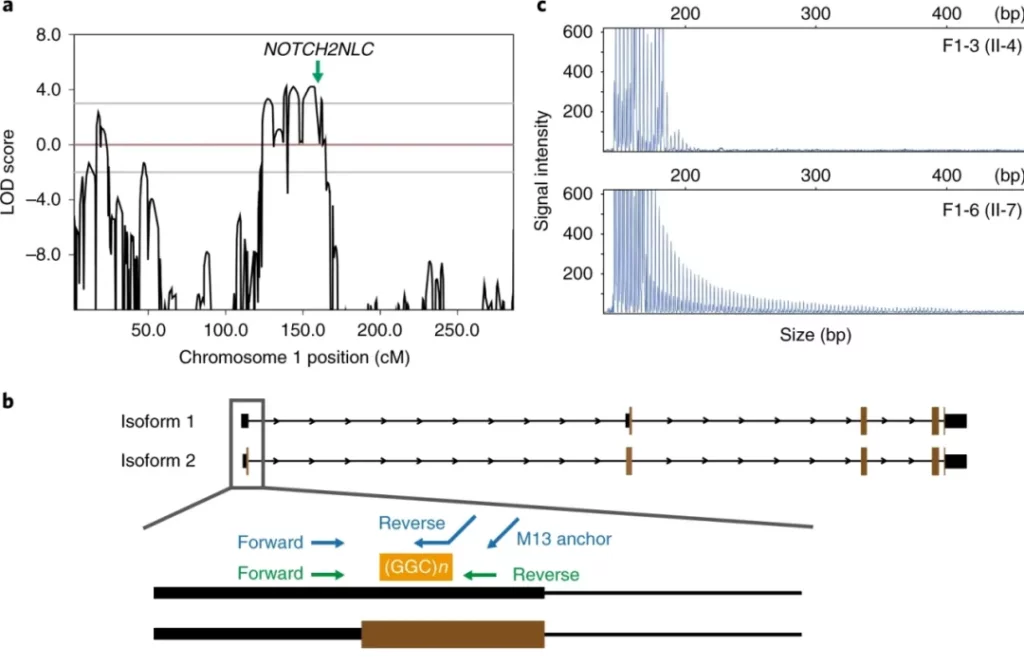
研究人员首先对家系1中的患病个体和正常个体进行了二代全基因组测序(WGS)或全外显子组测序(WES),并通过连锁分析将致病位点定位在1p36.31-p36.22 (chr 1: 6218354-9719813 [maximum LOD 2.32])约3.5-Mb 区域和1p22.1-q21.3 (chr 1: 94670784-152323132 [maximum LOD 4.21])约58.1-Mb区域内,但在二代WGS和WES测序数据中并没有发现致病性的SNPs或CNVs。
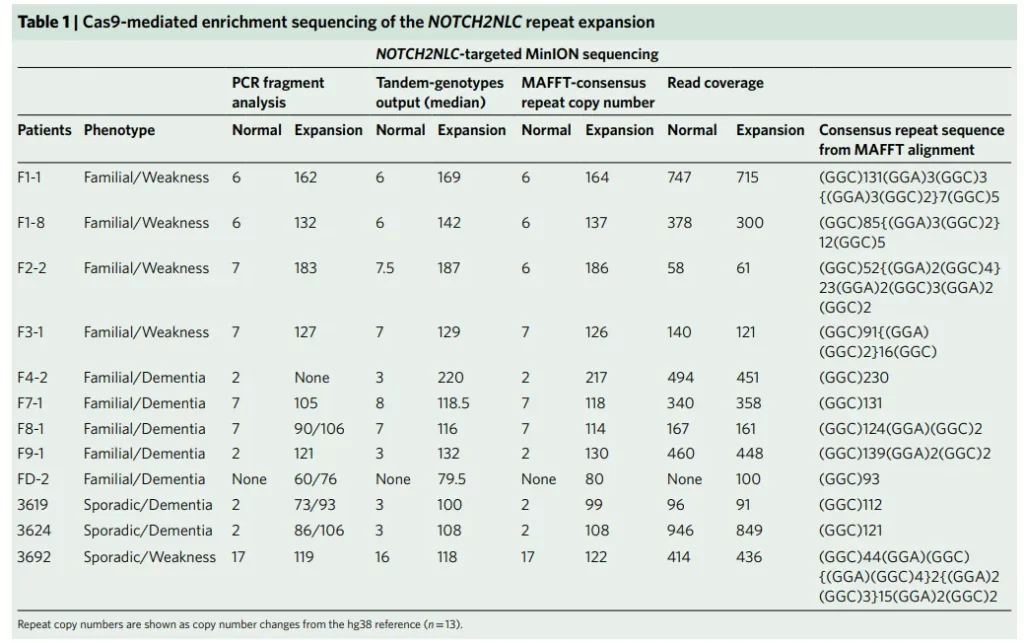
随后研究人员利用三代测序平台对来自8个家庭的13个患者和4例正常个体进行了全基因组测序,其中,除“F1-6”患者用PacBio RSII平台检测外,其余个体均用Oxford Nanopore PromethION平台进行检测。测序结果表明,13个患者在chr1:149390802-149390842处都有GGC重复扩增,该扩增序列位于NOTCH2NLC基因(NM_001364012)的5’UTR区域,定位在58.1-Mb的连锁区域内。对重复扩增区域进行进一步分析发现,除GGC重复单元外,重复序列中可能还包含GGA重复单元,这是由于在家系1、2和3患者的测序数据中都有reads支持GGA重复单元的存在,且F1-6的PacBio reads中也存在GGA重复单元。
NOTCH2NLC位于1q21.1,是人类特有的三个NOTCH2NL基因(NOTCH2NLA、NOTCH2NLB和NOTCH2NLC)之一,在包括神经胶质细胞、星形胶质细胞和小胶质细胞在内的多种神经细胞中高度表达,被认为参与了人类大脑皮层的进化。NOTCH2NL基因间具有高度的序列相似性,且多为GC-rich区域,因此难以用二代测序发等方法进行分析。而长读长测序技术,对于分析此类同源和重复区域具有很大优势。本研究还发现利用Nanopore PromethION 平台对NIID患者进行全基因组测序,即使在低覆盖度(~8X)下,也能检测到NOTCH2NLC基因中的重复扩张序列。
研究人员还利用Nanopore测序数据还分析了GGC重复扩张序列的甲基化水平,发现NOTCH2NLC的甲基化水平在GGC重复扩增区域与未扩增区域之间并无差异。因此认为由GGC扩增导致的CpG甲基化导致NOTCH2NLC功能丧失的可能性较小。
参考文献
1.Wu, Z., Jiang, Z., Li, T. et al. Structural variants in the Chinese population and their impact on phenotypes, diseases and population adaptation. Nat Commun 12, 6501 (2021). https://doi.org/10.1038/s41467-021-26856-x
2.Sone, J., Mitsuhashi, S., Fujita, A. et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 51, 1215–1221 (2019). https://doi.org/10.1038/s41588-019-0459-y

电话:027-870502099
邮箱:sales@rxbio.cc
地址:武汉市东湖高新区高新二路388 号
光谷生物医药加速器 18 栋 1-2层
单细胞多组学 空间转录组
三代测序 功能基因组
表观遗传学 互作组学
单细胞大数据 数据深度挖掘

欢迎关注公众号「瑞兴生物」