
解决核心痛点:长读长为何至关重要?
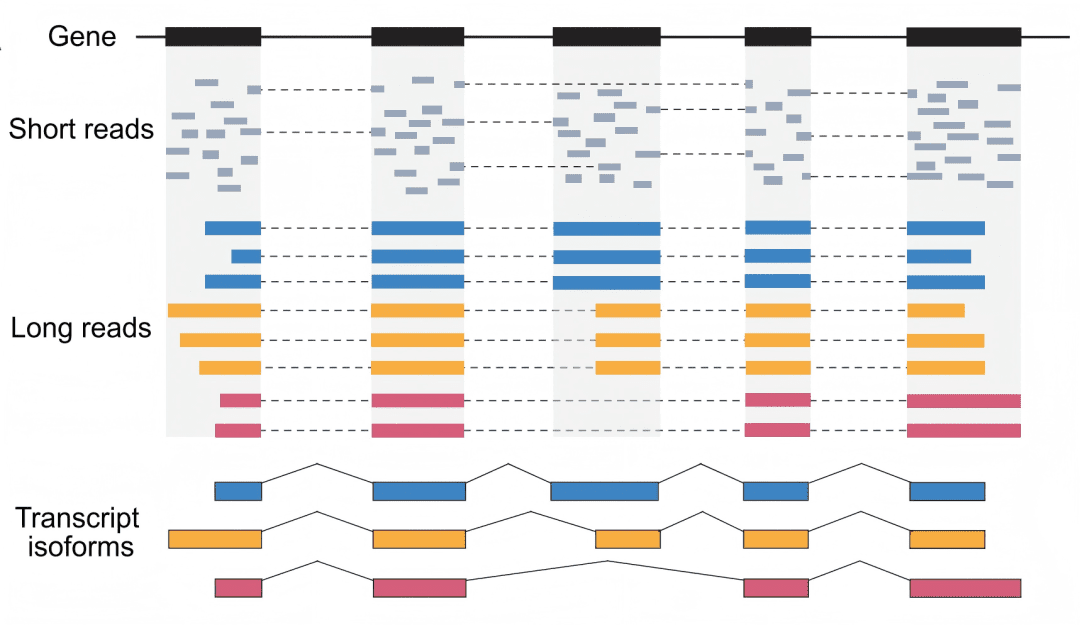
长读长技术解决的三大核心问题,这些都是研究人员常遇到的瓶颈:
1. 异构体难题:>95%的多外显子基因存在可变剪接,产生功能各异的异构体。短读长数据难以准确重建这些全长异构体。
2. 重复序列困境:约占基因组50%的重复序列(如转座子TE)在癌症中被异常激活,但短读长读段无法唯一比对,导致信息丢失。
3. 相位(Phasing)缺失:短读长无法确定同一个体细胞中,多个变异(如SNP和剪接事件)是否发生在同一条染色体上,难以进行等位基因特异性分析。
长读长测序一次性读取数千至数万个碱基,像“阅读整段句子”一样直接解析完整RNA分子,从根本上突破了这些限制。
核心应用领域与经典案例
应用一:精准解析癌症转录组复杂性,发现新型诊断标志物与治疗靶点
1. 发现肿瘤特异性异构体(Tumor-Specific Isoforms)
驱动肿瘤发生发展的往往是特定异构体,而非整个基因的表达变化。这些异构体可作为高特异性生物标志物或新抗原来源。

Chen et al. (2019) 使用PacBio长读长测序在肝细胞癌中鉴定出 ARHGEP2, DEK, ADRM1, 和 CD44 基因的新型转录本。这些异构体在肿瘤中高表达,但在正常肝脏组织中几乎不表达,是具有巨大潜力的诊断标志物。
2. 揭示耐药机制与精准用药
直接检测融合基因或关键致癌基因(如PIK3CA)的完整突变谱和单倍型,指导靶向治疗。

Cavelier et al. (2015) 利用长读长测序在单分子水平解析了慢性粒细胞白血病(CML) 患者中BCR-ABL1融合基因的突变复合体和剪接异构体,揭示了酪氨酸激酶抑制剂(TKI)耐药的分子机制 。

Vasan et al. (2019) 发现乳腺癌中PIK3CA基因的双突变若发生在同一等位基因上(顺式构型),会增强致癌性并使癌细胞对PI3Kα抑制剂更敏感,这一发现依赖长读长测序进行单倍型解析 。
3.全面鉴定致癌基因融合(Gene Fusions)
长读长不仅能发现融合,还能直接揭示断点处的精确序列和融合后的全长异构体结构,这是发现“双跳融合”等复杂事件的关键。

Namba et al. (2021) 在乳腺癌中发现了由复杂结构变异导致的双跳融合转录本(double-hop fusions)。

Kiyose et al. (2022) 在HBV相关肝细胞癌中发现了涉及乙肝病毒与宿主转座子(TE) 的融合转录本 。

Liu et al. (2022)在人乳头瘤病毒(HPV)相关宫颈癌中发现高表达的HPV-人融合转录本,例如HPV16 E6*I-E7-E1SD880-人基因,是宫颈癌发生的关键驱动因素,其可引发E6*I和E7的过度表达,并破坏肿瘤抑制基因CMAHP、TP63和P3H2的转录。
4. 挖掘免疫治疗新靶点(Neoantigens)
由异常剪接、TE激活等RNA失调产生的新抗原是癌症免疫治疗的宝贵资源。长读长测序是系统发现这类新抗原的唯一有效手段。

Oka et al. (2021) 对非小细胞肺癌(NSCLC) 进行全长转录组测序,旨在发现由异常剪接产生的、可能作为新抗原的转录本 。

Wang et al. (2023) 在乳腺癌细胞系中发现了肿瘤抑制基因的特异性异构体富集无义介导的mRNA降解(NMD)现象,提示了一种常见的RNA水平抑癌机制 。
应用二:揭示复杂疾病(神经/免疫)的转录调控新机制
1. 发现疾病相关异构体切换(Isoform Switching)
在总体基因表达不变的情况下,异构体比例的变化可能是疾病的核心分子表型。

Heberle et al. (2024) 对阿尔茨海默病(AD) 患者和对照的额叶皮层进行深度纳米孔测序,发现99个差异表达的转录本,其所属基因的整体表达并未发生变化。这凸显了异构体水平分析对于理解神经退行性疾病机制的必要性 。
2. 解析神经精神疾病风险基因的极端复杂性
许多GWAS发现的疾病风险基因位于非编码区,其功能机制不明。长读长测序可发现这些区域表达的全新转录本,将基因型与表型联系起来。

Hardwick et al. (2019) 结合靶向捕获与纳米孔测序,发现与神经精神性状相关的基因座中,62% 表达了之前未注释的多外显子转录本 。

Clark et al. (2020) 揭示了精神疾病风险基因 CACNA1C 在人脑中的异常复杂剪接模式 。
3. 增强罕见变异致病性解读
利用全长转录本图谱重新评估罕见遗传变异,可为大量“意义不明”的变异提供致病性证据,特别是在神经发育疾病中。

Patowary et al. (2024) 绘制了人新皮层发育的全长转录组图谱,发现了超过20万个异构体。利用该图谱,他们重新评估并优先考虑了数千个罕见变异,将其与自闭症、智力残疾等疾病联系起来,极大增强了遗传发现的解读能力 。
应用三:提升罕见遗传病的分子诊断率
1. 直接验证剪接突变致病性
对DNA测序发现的疑似剪接位点突变或深在内含子突变,进行RNA水平的功能验证,是确诊的关键步骤。

Sedaghat-Hamedani et al. (2022) 利用纳米孔测序在一个大型扩张型心肌病家系中明确了 LMNA 基因剪接变异的致病性 。

Dainis et al. (2019) 在 MYBPC3 基因中揭示了剪接位点变异导致的转录本异常 。

Chandrasekhar et al. (2024) 利用靶向长读长测序系统研究了 USH2A 基因中的剪接缺陷,成功破解了由深在内含子变异和“意义不明”变异引起的多种简单和复杂错误剪接模式,为患者提供了明确诊断 。
2. 开发靶向诊断方案
针对已知致病基因 panel,开发靶向长读长RNA测序方法,可高效、同步检测多个基因的表达和剪接缺陷。

Schwenk et al. (2023) 开发了CAPLResq方法,用于林奇综合征(Lynch syndrome) 的诊断,通过超深度的长读长测序检测错配修复基因的RNA缺陷,展示了其临床应用的可行性 。
总结与展望
长读长RNA测序已不再是概念性技术,它正在全球顶尖的医学研究中产出实实在在的成果。它为解决以下临床科研问题提供了强大工具:
● 当DNA测序找不到明确答案时 → 寻找RNA水平的异常(剪接错误、等位基因特异性表达)。
● 当基因表达谱结果看似“正常”时 → 挖掘关键的异构体失衡。
● 当寻求更高特异性的生物标志物时 → 发现疾病特异性转录本(如来源于TE或基因融合的转录本)。
● 当探索免疫治疗新靶点时 → 系统扫描由RNA失调产生的新抗原。
随着测序成本下降、精度提升和数据分析工具的日益成熟,长读长RNA测序有望从目前的前沿研究工具,逐步转化为常规的分子诊断和转化研究平台。对于旨在课题上取得突破的临床科研工作者而言,率先布局和应用这一技术,无疑将占据领先优势。
参考文献
-
Ament IH, DeBruyne N, Wang F, Lin L. Long-read RNA sequencing: A transformative technology for exploring transcriptome complexity in human diseases. Mol Ther. 2025;33(3):883-894. doi:10.1016/j.ymthe.2024.11.025
-
Chen H., Gao F., He M., Ding X.F., Wong A.M., Sze S.C., Yu A.C., Sun T., Chan A.W.H., Wang X., Wong N. Long-Read RNA Sequencing Identifies Alternative Splice Variants in Hepatocellular Carcinoma and Tumor-Specific Isoforms. Hepatology. 2019;70:1011–1025. doi: 10.1002/hep.30500.
-
Cavelier L., Ameur A., Häggqvist S., Höijer I., Cahill N., Olsson-Strömberg U., Hermanson M. Clonal distribution of BCR-ABL1 mutations and splice isoforms by single-molecule long-read RNA sequencing. BMC Cancer. 2015;15:45. doi: 10.1186/s12885-015-1046-y.
-
Vasan N., Razavi P., Johnson J.L., Shao H., Shah H., Antoine A., Ladewig E., Gorelick A., Lin T.-Y., Toska E., et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science. 2019;366:714–723. doi: 10.1126/science.aaw9032.
-
Namba S., Ueno T., Kojima S., Kobayashi K., Kawase K., Tanaka Y., Inoue S., Kishigami F., Kawashima S., Maeda N., et al. Transcript-targeted analysis reveals isoform alterations and double-hop fusions in breast cancer. Commun. Biol. 2021;4:1320. doi: 10.1038/s42003-021-02833-4.
-
Kiyose H., Nakagawa H., Ono A., Aikata H., Ueno M., Hayami S., Yamaue H., Chayama K., Shimada M., Wong J.H., Fujimoto A. Comprehensive analysis of full-length transcripts reveals novel splicing abnormalities and oncogenic transcripts in liver cancer. Plos Genet. 2022;18 doi: 10.1371/journal.pgen.1010342.
-
Oka M., Xu L., Suzuki T., Yoshikawa T., Sakamoto H., Uemura H., Yoshizawa A.C., Suzuki Y., Nakatsura T., Ishihama Y., et al. Aberrant splicing isoforms detected by full-length transcriptome sequencing as transcripts of potential neoantigens in non-small cell lung cancer. Genome Biol. 2021;22:9. doi: 10.1186/s13059-020-02240-8.
-
Wang F., Xu Y., Wang R., Zhang B., Smith N., Notaro A., Gaerlan S., Kutschera E., Kadash-Edmondson K.E., Xing Y., Lin L. TEQUILA-seq: a versatile and low-cost method for targeted long-read RNA sequencing. Nat. Commun. 2023;14:4760. doi: 10.1038/s41467-023-40083-6.
-
Aguzzoli Heberle B., Brandon J.A., Page M.L., Nations K.A., Dikobe K.I., White B.J., Gordon L.A., Fox G.A., Wadsworth M.E., Doyle P.H., et al. Mapping medically relevant RNA isoform diversity in the aged human frontal cortex with deep long-read RNA-seq. Nat. Biotechnol. 2024 doi: 10.1038/s41587-024-02245-9.
-
Hardwick S.A., Bassett S.D., Kaczorowski D., Blackburn J., Barton K., Bartonicek N., Carswell S.L., Tilgner H.U., Loy C., Halliday G., et al. Targeted, High-Resolution RNA Sequencing of Non-coding Genomic Regions Associated With Neuropsychiatric Functions. Front. Genet. 2019;10:309. doi: 10.3389/fgene.2019.00309.
-
Clark M.B., Wrzesinski T., Garcia A.B., Hall N.A.L., Kleinman J.E., Hyde T., Weinberger D.R., Harrison P.J., Haerty W., Tunbridge E.M. Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Mol. Psychiatry. 2020;25:37–47. doi: 10.1038/s41380-019-0583-1.
-
Patowary A., Zhang P., Jops C., Vuong C.K., Ge X., Hou K., Kim M., Gong N., Margolis M., Vo D., et al. Developmental isoform diversity in the human neocortex informs neuropsychiatric risk mechanisms. Science. 2024;384 doi: 10.1126/science.adh7688.
-
Sedaghat-Hamedani F., Rebs S., Kayvanpour E., Zhu C., Amr A., Müller M., Haas J., Wu J., Steinmetz L.M., Ehlermann P., et al. Genotype Complements the Phenotype: Identification of the Pathogenicity of an LMNA Splice Variant by Nanopore Long-Read Sequencing in a Large DCM Family. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms232012230.
-
Dainis A., Tseng E., Clark T.A., Hon T., Wheeler M., Ashley E. Targeted Long-Read RNA Sequencing Demonstrates Transcriptional Diversity Driven by Splice-Site Variation in MYBPC3. Circ. Genom Precis Med. 2019;12 doi: 10.1161/CIRCGEN.119.002464.
-
Chandrasekhar S., Lin S., Jurkute N., Oprych K., Estramiana Elorrieta L., Schiff E., Malka S., Wright G., Michaelides M., Mahroo O.A., et al. Investigating Splice Defects in USH2A Using Targeted Long-Read Sequencing. Cells. 2024;13:1261. doi: 10.3390/cells13151261.
-
Schwenk V., Leal Silva R.M., Scharf F., Knaust K., Wendlandt M., Häusser T., Pickl J.M.A., Steinke-Lange V., Laner A., Morak M., et al. Transcript capture and ultradeep long-read RNA sequencing (CAPLRseq) to diagnose HNPCC/Lynch syndrome. J. Med. Genet. 2023;60:747–759. doi: 10.1136/jmg-2022-108931.