

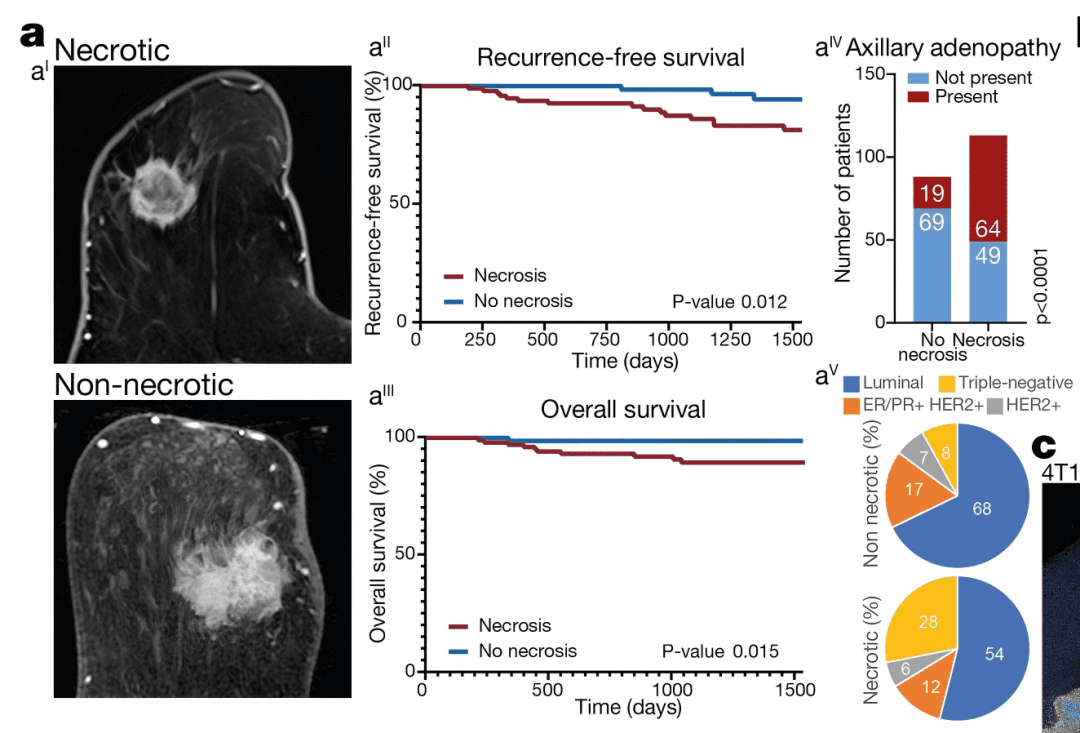
Extended Data Fig. 1a
NETs(中性粒细胞胞外陷阱)是中性粒细胞释放的DNA-蛋白质网状结构,原本用于捕获病原体,却在肿瘤中“失控”:研究发现,肿瘤会诱导出一类特殊的中性粒细胞亚群——Ly6G⁺Ly6Cᵀᴼᴸ(高表达Ly6G、低表达Ly6C),这类细胞无法像普通中性粒细胞那样迁移到炎症组织(即“不能外渗”),却能以2倍以上效率形成NETs(Fig. 4e)。
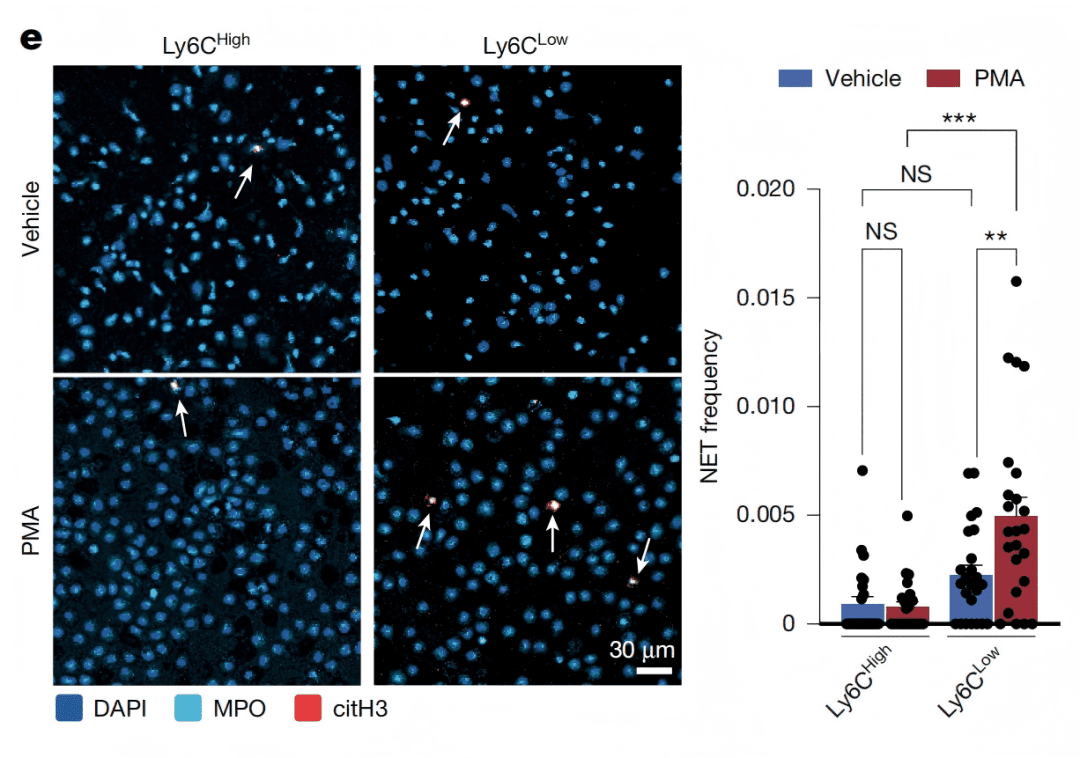
Fig. 4e
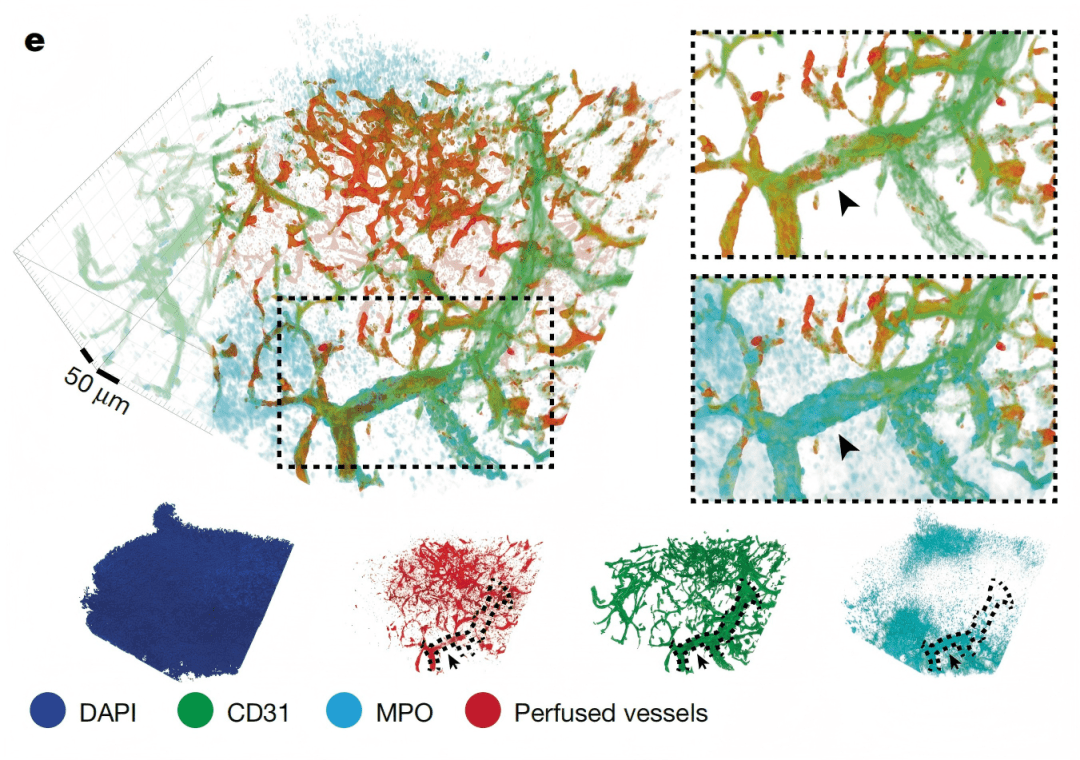
Fig. 1e

Fig. 1f
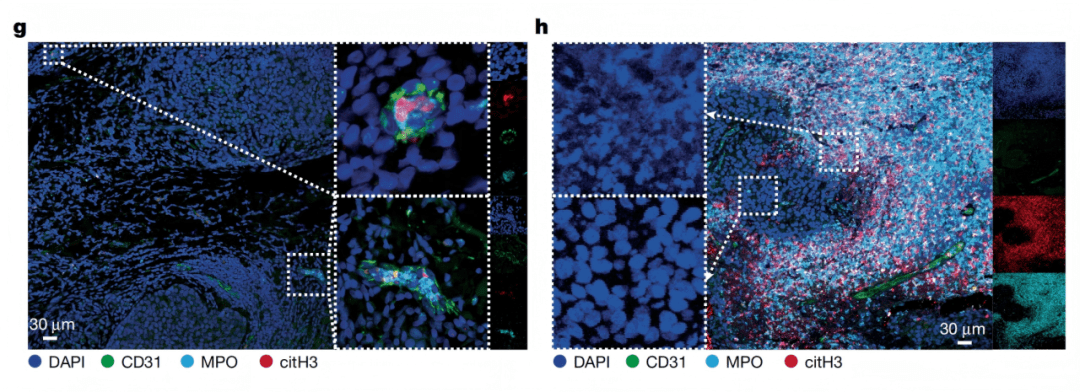
Fig. 1g–h
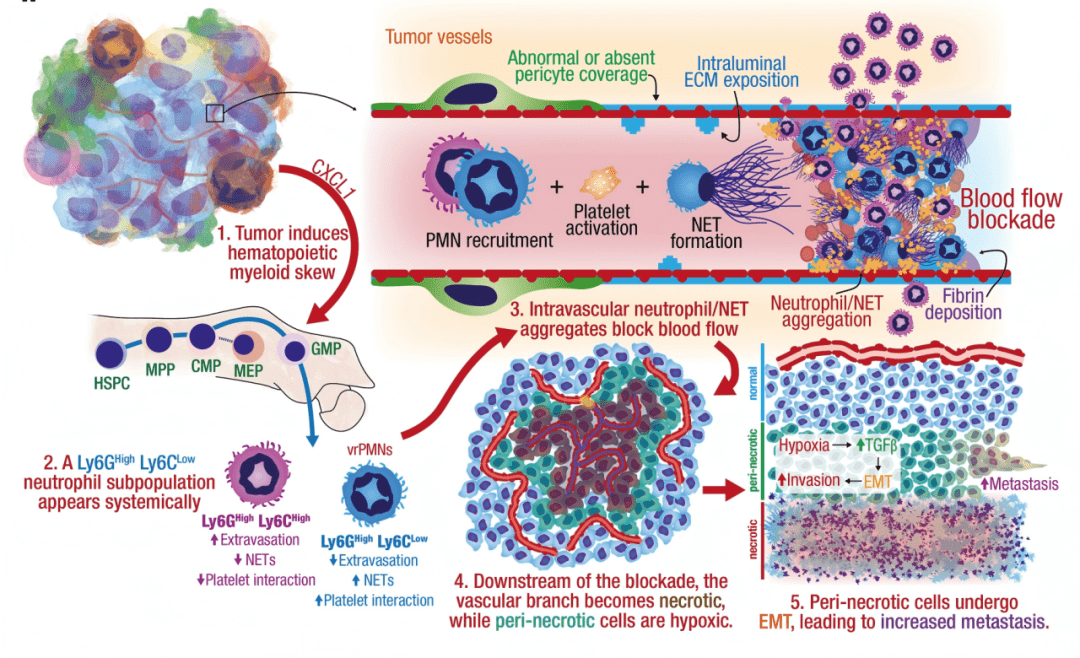
核心发现2:坏死旁区藏“转移种子”,TGFβ信号让癌细胞“学会游走
通过单细胞RNA测序(scRNA-seq)和空间转录组分析,团队发现:坏死旁区的癌细胞处于缺氧状态,且普遍激活“上皮间质转化(EMT)”程序——原本紧密连接的上皮型癌细胞(高表达E-钙粘蛋白,维持细胞粘附),转变为松散的间质型细胞(高表达波形蛋白,增强迁移能力),就像“解开束缚的细胞,获得了游走的能力”(Fig. 2b、i)。
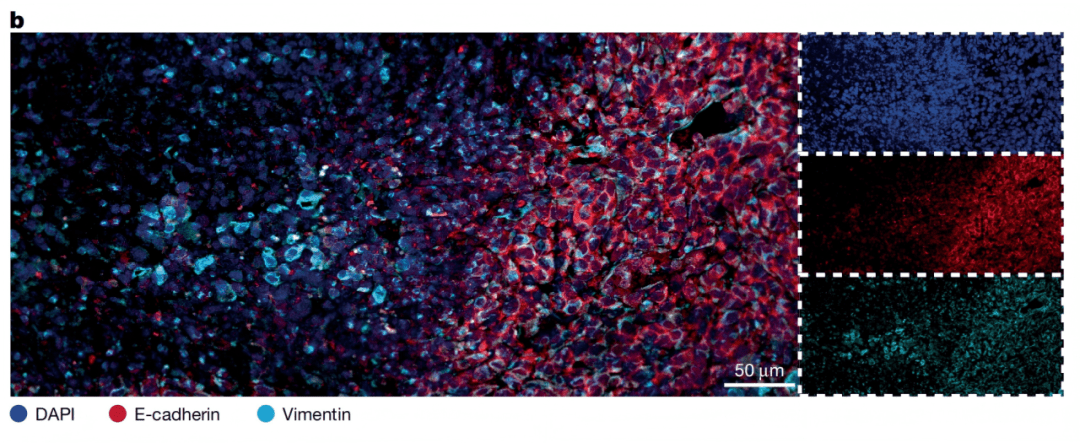
Fig. 2b
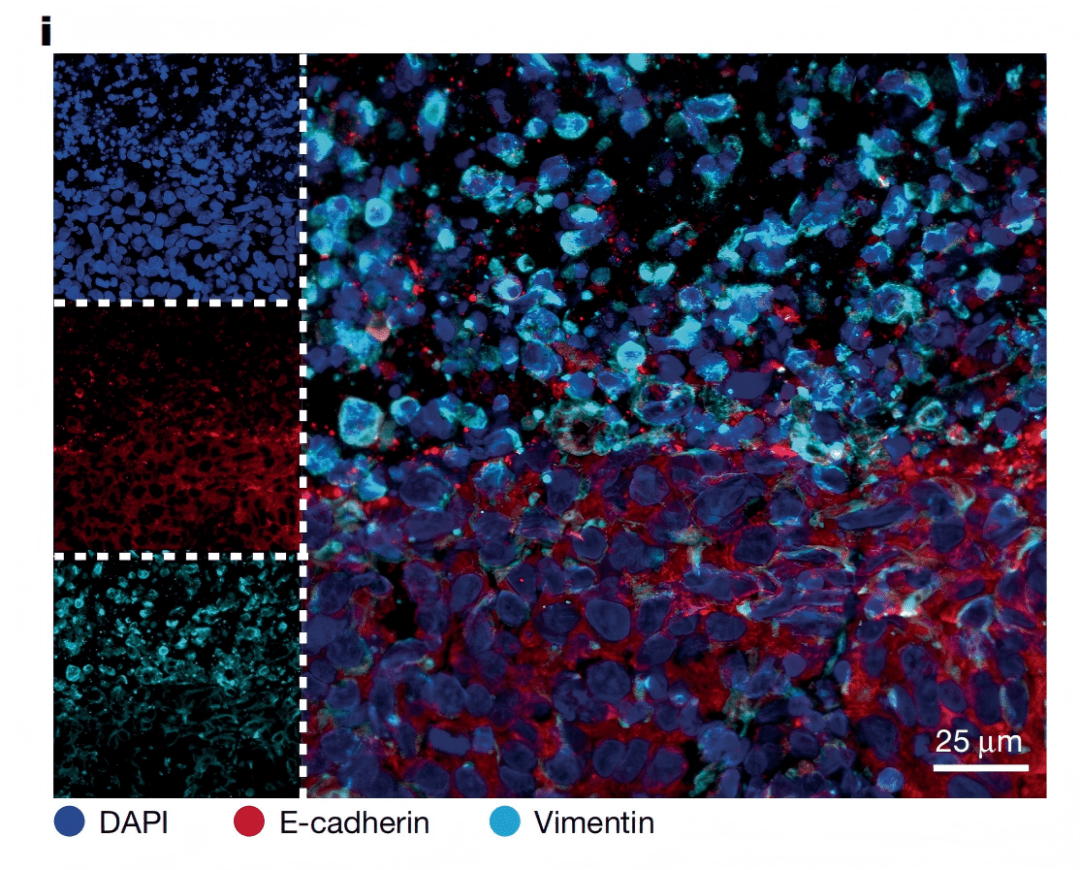
Fig. 2i
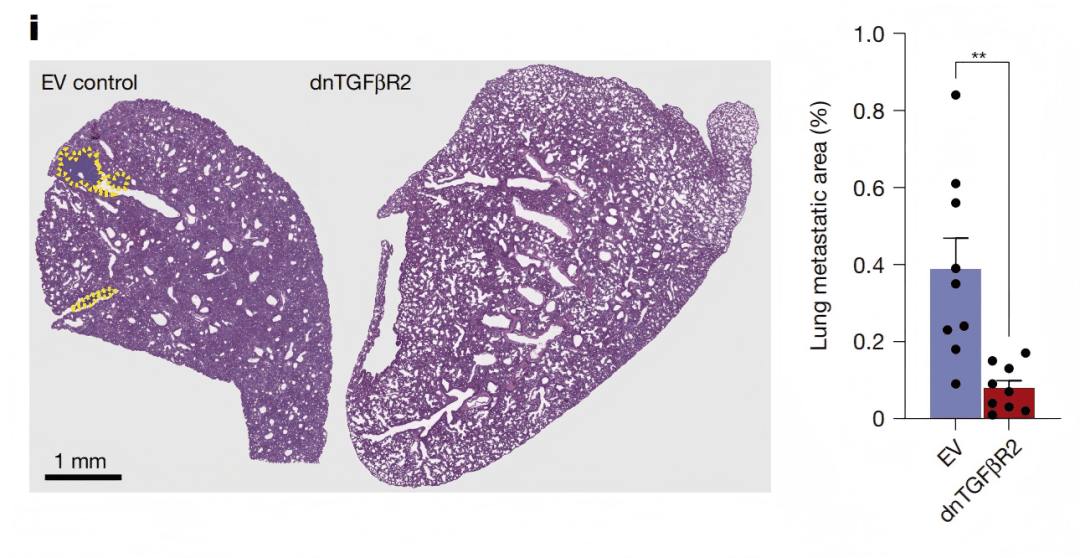
Fig. 3i
核心发现3:肿瘤“远程操控”造血系统,CXCL1是中性粒细胞“变节”的“信号弹”
对比不同肿瘤模型(4T1、LLC能诱导多形性坏死,MMTV-PyMT则不能)后,团队发现:能诱导坏死的肿瘤会高表达CXCL1(一种趋化因子),而CXCL1会通过两步“改造”中性粒细胞:
2. 诱导中性粒细胞“变节”:CXCL1还会促进中性粒细胞表型改变,诱导出“Ly6G⁺Ly6Cᵀᴼᴸ”亚群——这类细胞高表达CD11c(纤维蛋白受体)和ICAM-1(细胞粘附分子),更容易在血管内粘附、聚集并形成NETs(图4b)。
实验验证显示:当团队敲除4T1细胞的CXCL1基因后,小鼠外周血中“Ly6G⁺Ly6Cᵀᴼᴸ”中性粒细胞比例下降47%,肿瘤坏死面积减少58%,肺转移灶数量减少42%(Extended Data Fig. 10q–r)。这表明,CXCL1是肿瘤“策反”中性粒细胞、诱导坏死的关键“信号弹”。
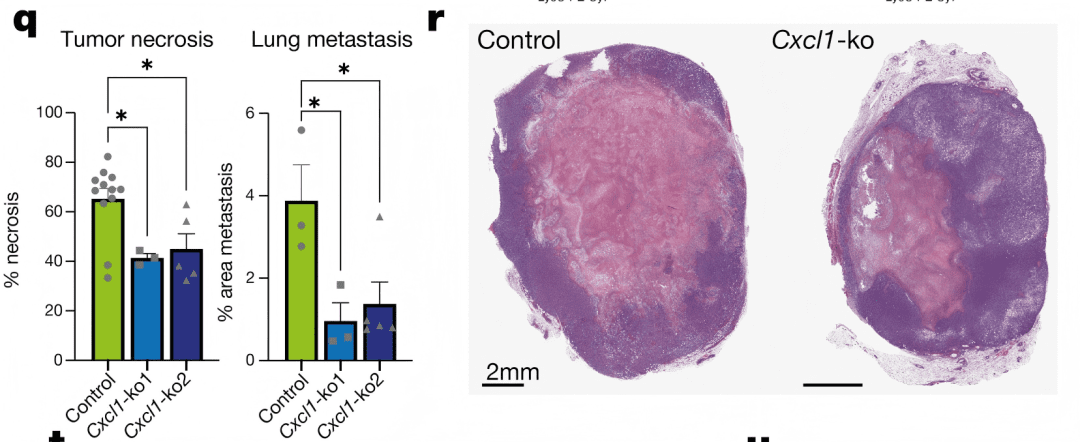
Extended Data Fig. 10q–r
临床突破:三种干预方式阻断“坏死-转移”轴,老药新用显潜力
● 肿瘤中NETs几乎完全消失,血管堵塞率下降82%;
● 坏死面积从对照组的60%降至5%以下(P=0.0025,Fig. 5d);
● 肺转移灶数量减少41%,转移面积减少38%(P=0.0144,Fig. 5f)。
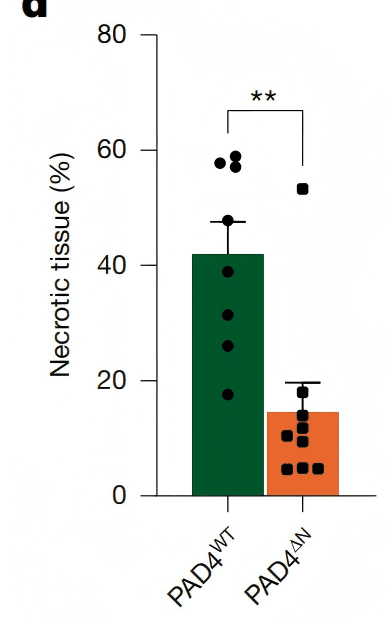
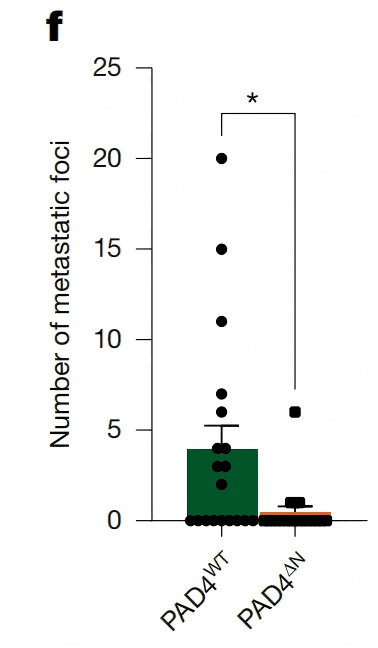
Fig. 5d、f
● 双硫仑(Disulfiram):FDA批准的酒精依赖治疗药物,此前被发现可抑制NET形成。给肿瘤小鼠喂食含双硫仑的饲料后(1g/kg diet),肿瘤坏死面积减少52%,癌细胞EMT特征消失,肺转移灶数量减少35%(Extended Data Fig. 12j-m),且不影响肿瘤大小或中性粒细胞总数,安全性良好。
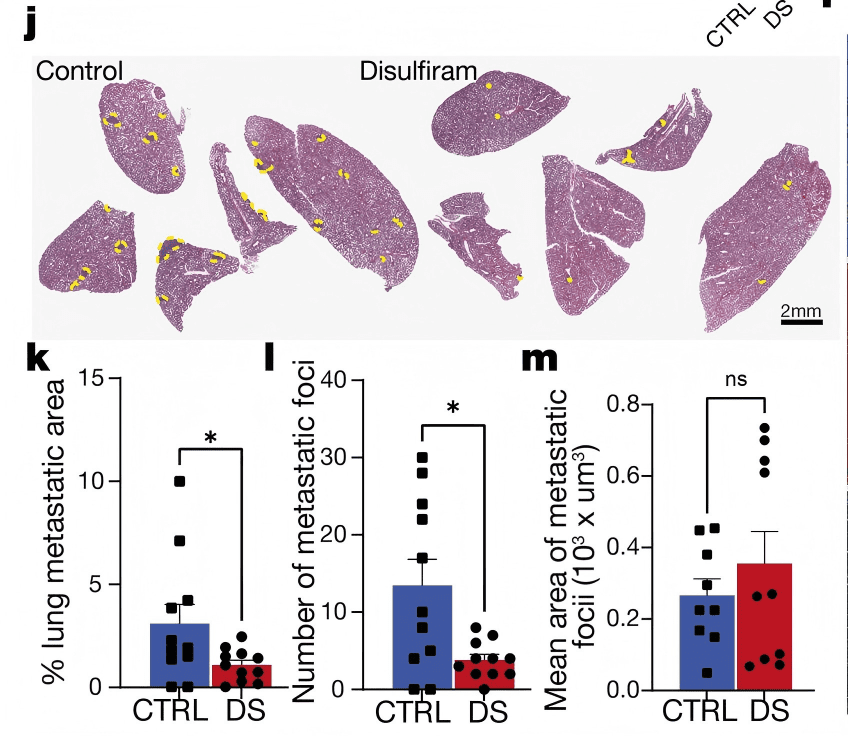
Extended Data Fig. 12j-m