

中文题目:利用nanoCAGE测序技术在基因组水平鉴定大豆转录起始位点
发表期刊:Scientific Data(Q1 IF=6.9)
发表时间:2025.10
DOI:10.1038/s41597-025-06003-7
欢迎咨询:027-87050299
研究背景
- 转录起始位点与启动子:基因表达的“开关”
基因表达的第一步是“转录”,而这个过程开始的精确位置被称为转录起始位点。它就像是基因的“启动按钮”。TSS上游紧邻的区域被称为启动子,是细胞调控机器识别并启动转录的关键区域。精确鉴定TSS是理解基因何时、何地以及如何被开启的基石,对于解析生长发育、疾病发生等生物学过程至关重要。
- 植物中的研究挑战与CAGE技术
尽管TSS如此重要,但在植物中,通过实验精确鉴定并绘制全基因组TSS图谱的研究仍然非常稀缺。传统的生物信息学预测往往不够准确。Cap Analysis of Gene Expression (CAGE) 是一种能够高通量、精准捕获TSS的先进技术。其原理是利用RNA分子5‘端特有的“帽子”结构,从而直接锁定转录的起始点,获得全基因组范围的TSS数据。本研究利用nanoCAGE 技术——一种所需RNA样本量更少、更灵敏的CAGE技术升级版。
- 启动子类型:“sharp” vs “broad”
根据TSS的分布模式,启动子可分为两种主要类型:“sharp”型 通常具有一个主导的TSS,常与组织特异性调控相关;而“broad”型 则包含一系列紧密相邻的TSS,常与组成型或动态调控的基因相关。解析启动子形状有助于深入理解基因的调控策略。
研究思路
第一阶段:数据获取与基础处理
采集大豆根与茎组织,利用nanoCAGE-seq技术构建文库并进行测序,直接捕获带有5‘帽子的完整mRNA转录本。随后对原始数据进行质控、去接头、去污染(如rRNA),并将高质量序列精准比对至大豆参考基因组。
第二阶段:TSS的系统鉴定与注释
将比对上的reads的5‘端位置定义为精确的TSS。随后,将相邻的TSS(距离在150 bp内)聚类为一个标签簇(TC),代表一个独立的转录单位。最后,将所有TSS和TCs与已知的基因注释进行关联,明确其基因组定位(如启动子区、基因间区等)。
第三阶段:启动子特征全局分析
对所有鉴定到的启动子(TCs)进行特征分析:首先,根据TSS的分布宽度,将其分为“sharp”和“broad”两种类型,并统计其分布比例。其次,提取核心启动子序列,通过生物信息学方法挖掘其保守的顺式作用元件,特别是对经典的TATA-box进行识别和富集度分析。
研究发现
通过nanoCAGE-seq技术,本研究在大豆根和茎组织中共精准鉴定了711,689个转录起始位点(CTSS)。这些TSS被进一步聚类为27,321个标签簇(TCs),对应于16,100个基因,首次在大豆中系统地绘制了高分辨率的全基因组TSS图谱,为后续启动子研究奠定了坚实基础。
2. 揭示大豆启动子以“sharp”类型为主导
研究对启动子形状进行了系统分析,发现大豆的启动子呈现出明显的“sharp”型主导特征。具体而言,在所有鉴定到的TCs中,66.36%为“sharp”型,而仅有33.64%为“broad”型。这一比例特征与玉米类似,但不同于拟南芥,表明启动子结构可能具有物种特异性。
3. 鉴定出核心启动子区富集的TATA-box motif
通过对核心启动子序列(-50 bp 到 +50 bp)进行motif分析,研究发现在TSS上游约30 bp(-35至-25区间)处显著富集一个TA-rich motif,该序列即为经典的TATA-box核心元件。进一步分析显示,该 motif 在 46% 的sharp启动子 和 40% 的broad启动子 中出现,证实了TATA-box在大豆转录起始中的重要作用。
4. 验证数据高质量与高可靠性
技术验证表明,本研究产生的nanoCAGE-seq数据质量高且可靠。两个生物学重复之间的Pearson相关系数极高(根:0.98;茎:0.97),显示出优异的可重复性。同时,CAGE标签的分布符合预期的幂律分布,且绝大多数(86.24%)CTSS位于已知基因的启动子区域,共同证明了本数据集的高准确度和可靠性。
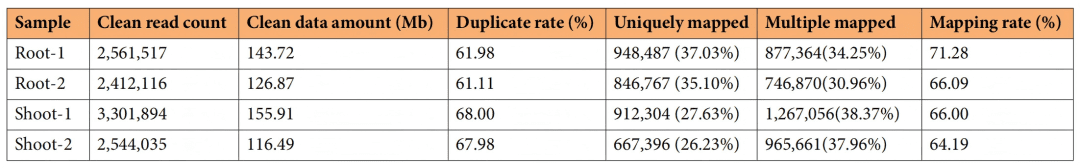
所有样本均获得百万级高质量测序 reads,总体比对率在64%-71%之间,证明了数据质量可靠,可用于下游分析。
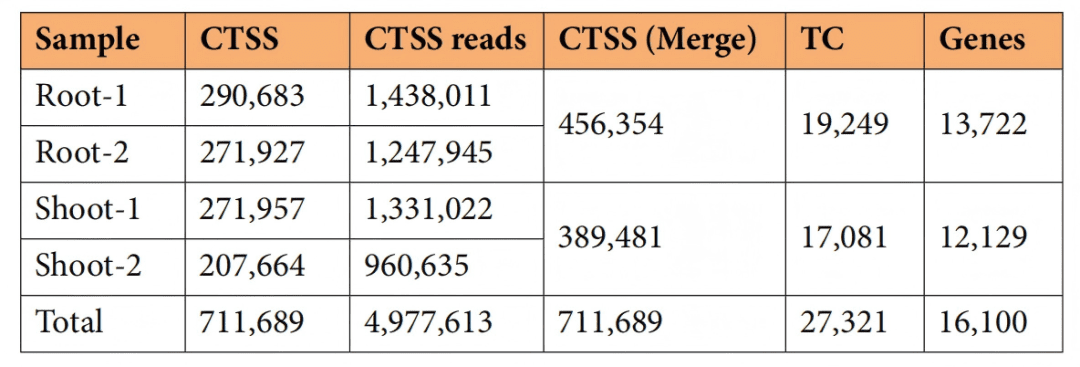
本研究共鉴定出超过71万个CTSS,聚类为2.7万多个TC,覆盖了约1.6万个基因,系统描绘了大豆的启动子景观。
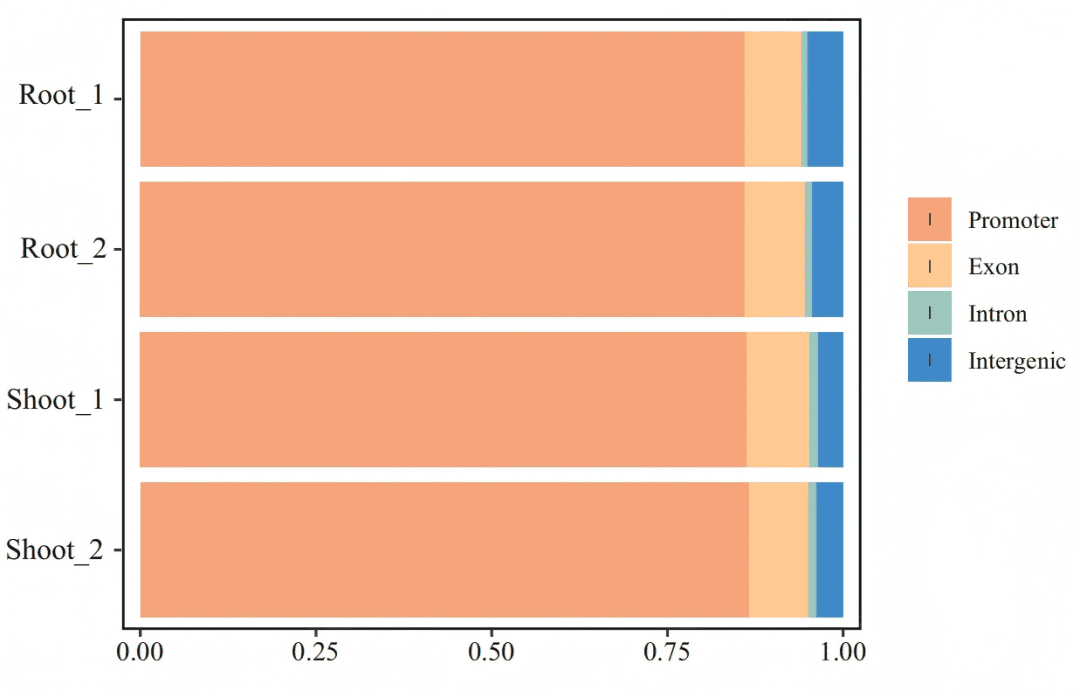
绝大多数(86%)的CTSS位于启动子区域,与预期相符,证明了实验技术能精准定位转录起始位点。
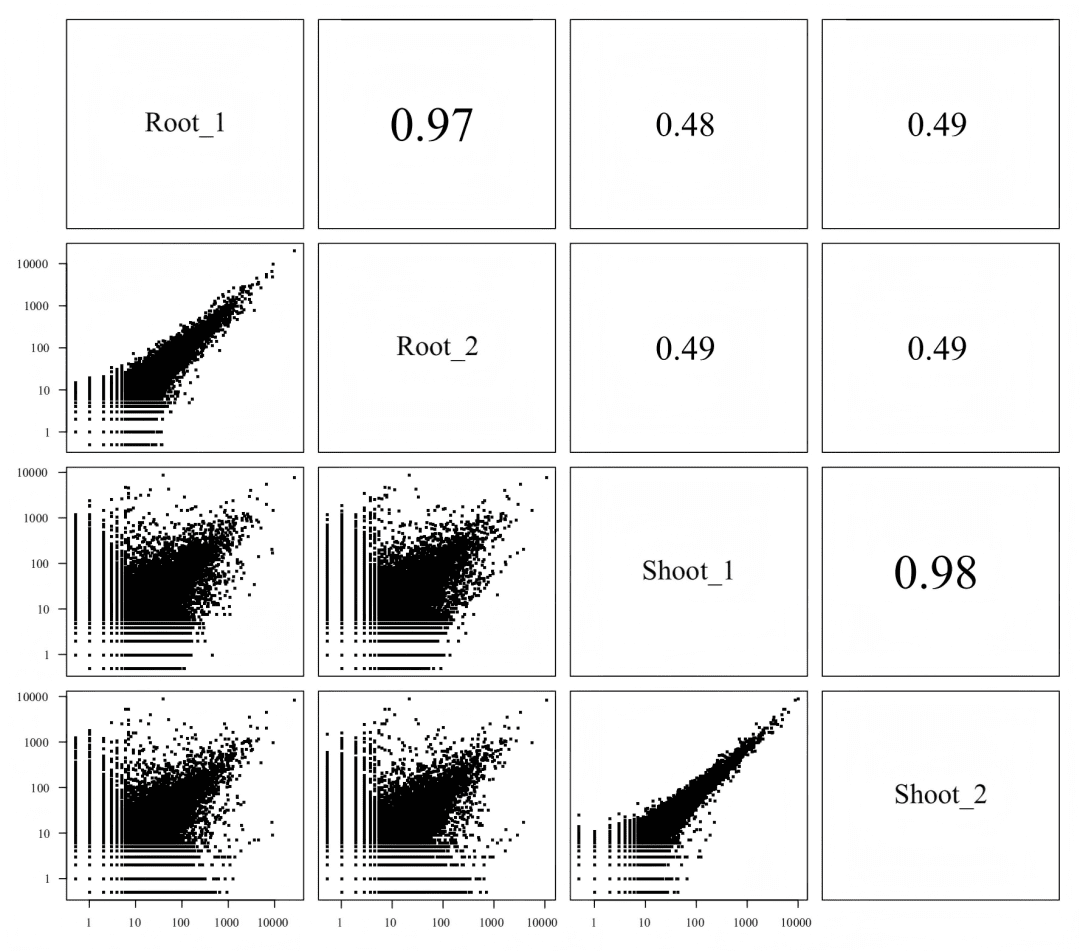
生物学重复间相关性极高(r>0.97),而不同组织间相关性较低,证明了数据的高重复性和组织特异性。
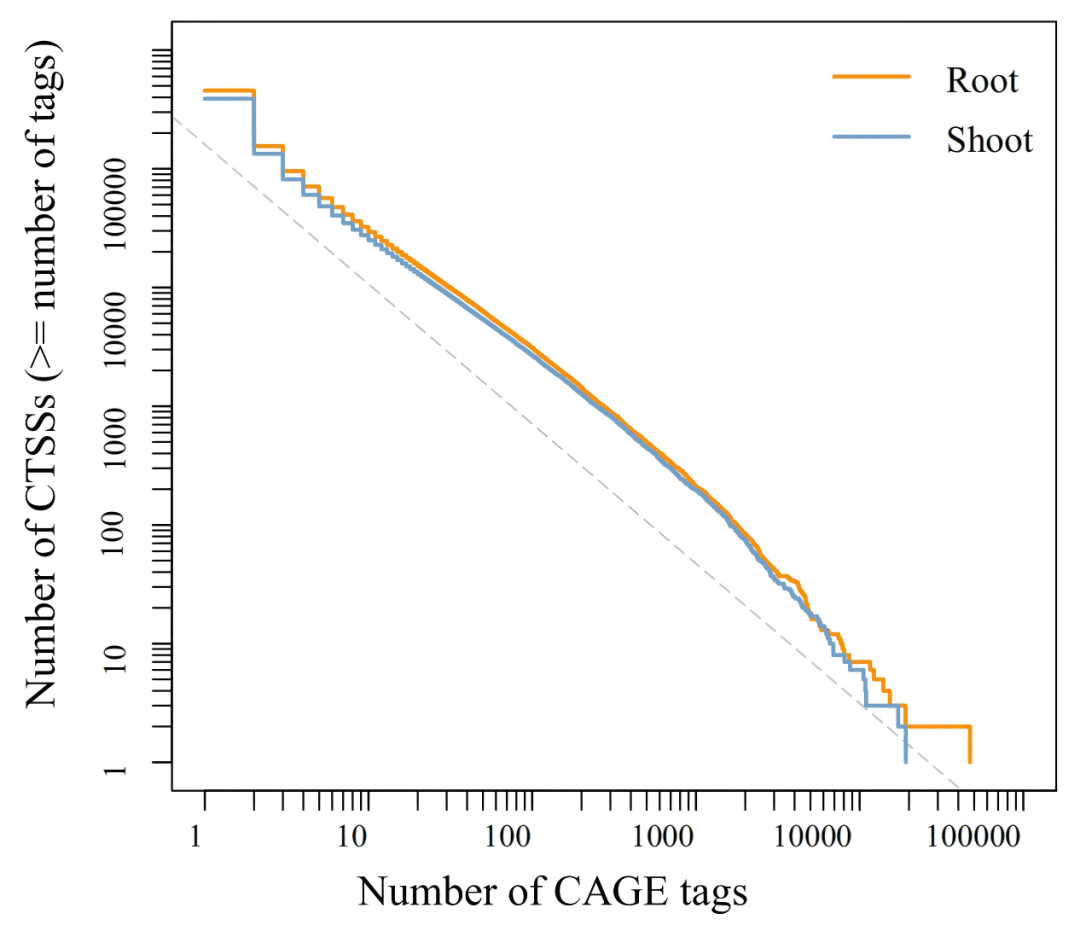
数据分布呈近似直线,符合高质量CAGE数据特有的幂律分布,再次验证了数据质量。
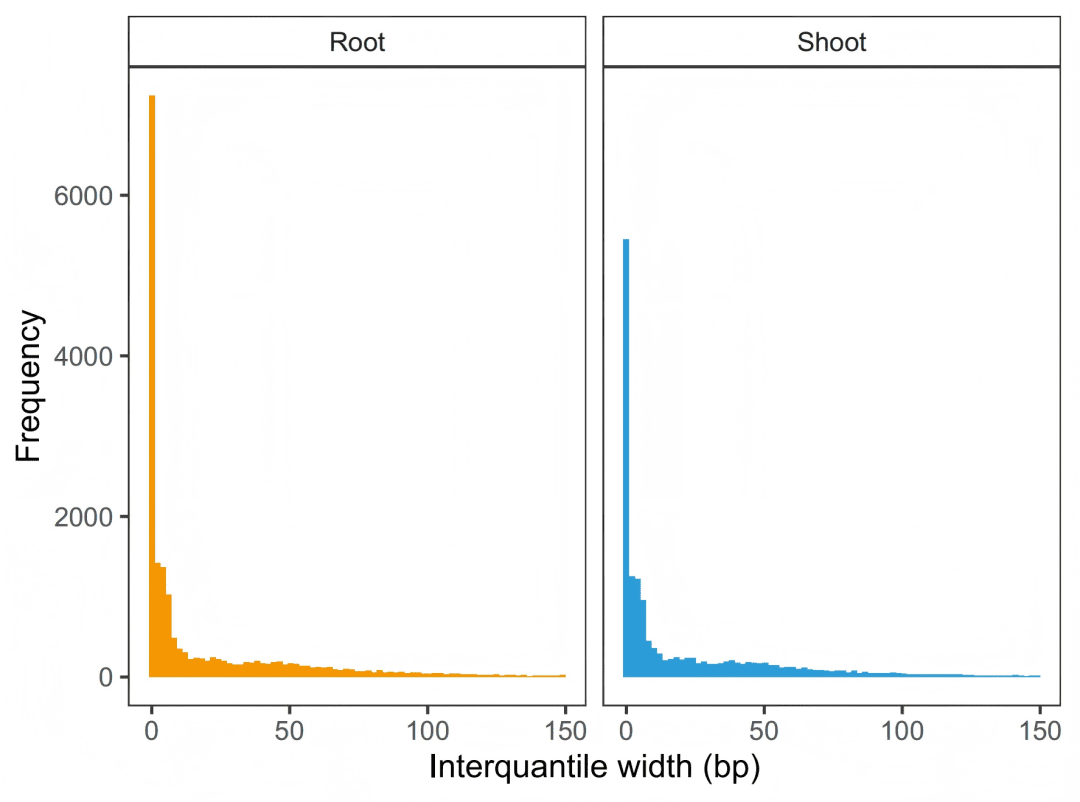
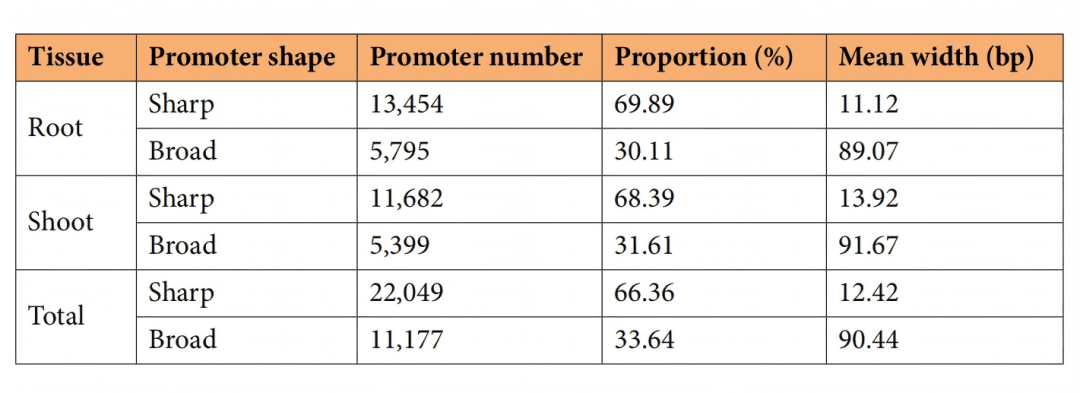
大豆启动子以“sharp”型为主(占总数的66%以上),且在根和茎组织中的分布比例非常接近,表明这是大豆基因组的一个固有特征。
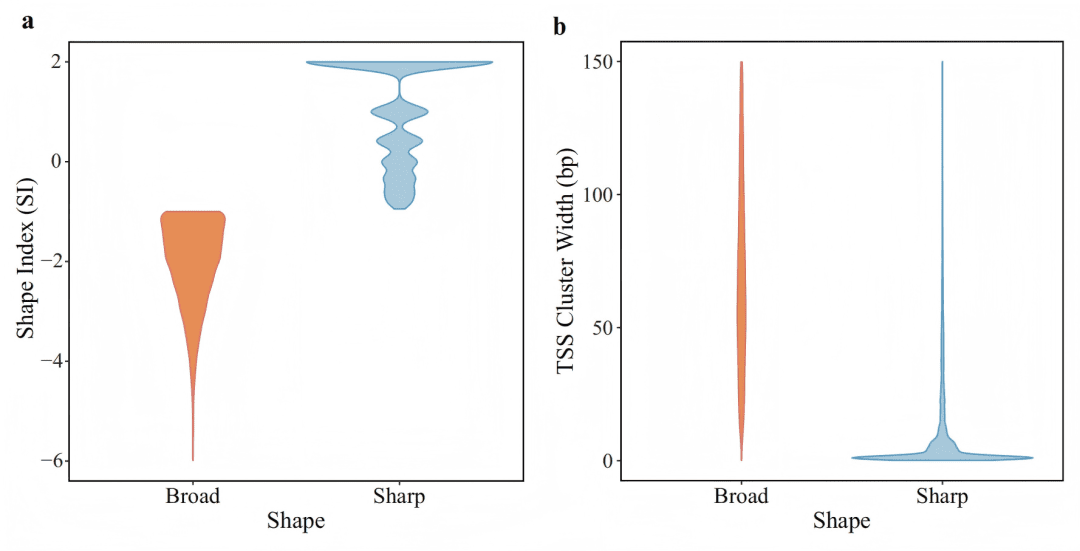
清晰地将启动子定量分为“sharp”(分布集中、宽度窄)和“broad”(分布分散、宽度大)两类,并显示“sharp”型占大多数。
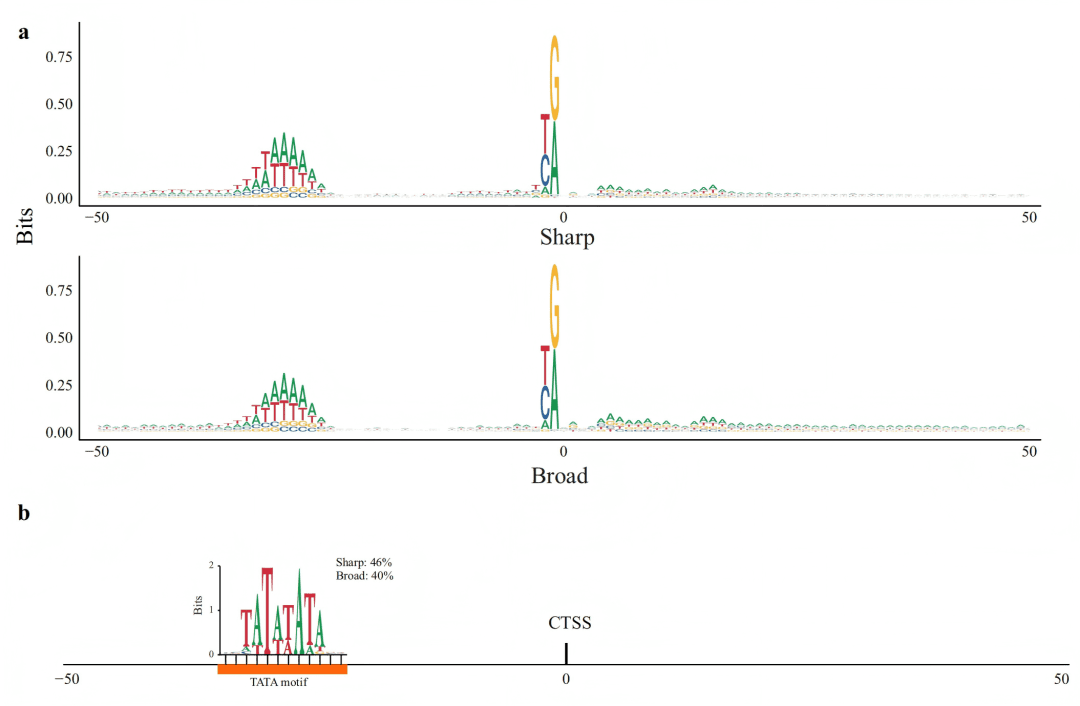
在TSS上游约30 bp处显著富集TA motif(即TATA-box),且其在“sharp”型启动子中更为常见(46%),揭示了大豆核心启动子的关键序列特征。
Fang, Weiwei, et al. “Genome Level Identification of Transcription Start Sites by NanoCAGE Sequence in Soybean.” Scientific Data, vol. 12, 2025, article 1723. Nature, https://doi.org/10.1038/s41597-025-06003-7.