
一项发表于《Nature Genetics》的重磅研究,利用单细胞多组学(scRNA-seq、snATAC-seq)与空间转录组(Stereo-seq),为我们精准“锁定”了元凶——一个名为 GPR116+周细胞 的全新细胞亚群。研究发现,这群细胞不仅是PRRX1驱动的“转移引擎”,通过EGFL6-integrin β1轴强力驱动癌细胞侵袭;更是“免疫哨卡的构筑者”,直接塑造抑制性免疫微境,导致T细胞耗竭。研究不仅从空间原位揭示了ESCC的恶性进化机制,更直接指向了血清EGFL6这一无创诊断标志物,以及靶向整合素β1联合免疫检查点抑制剂的协同治疗新策略,为克服食管癌转移与免疫耐药开辟了令人振奋的新路径。

中文题目:食管鳞状细胞癌的单细胞多组学与空间分析揭示GPR116+周细胞在癌症转移中的免疫抑制角色
发表期刊:Nature Genetics(Q1,IF=29)
DOI:https://doi.org/10.1038/s41588-025-02341-9
研究背景
食管鳞癌是中国高发的恶性肿瘤,其致命主因是肿瘤转移。多数患者确诊时已发生转移,导致五年生存率极低(约20%)。尽管免疫疗法等新兴手段出现,但疗效仍不理想,揭示其内在抵抗机制是当务之急。
肿瘤微环境(TME):转移的“帮凶”
肿瘤并非由癌细胞独立构成,它身处一个复杂的生态系统——肿瘤微环境(TME)。TME由多种基质细胞(如成纤维细胞、免疫细胞、血管周细胞等)和细胞外成分共同构成。传统观点认为,转移主要取决于癌细胞自身的特性;但现在我们认识到,TME中各种细胞与癌细胞的动态相互作用,是启动和促进转移过程的关键。
周细胞:被忽视的关键角色
周细胞是包裹在毛细血管壁外的细胞,传统功能被认为是维持血管稳定性。在肿瘤中,周细胞的表现非常复杂且存在异质性(即存在功能不同的亚群)。它们既能抑制肿瘤,也能促进其发展。然而,由于周细胞在组织中含量稀少,其具体的亚群分类、功能以及在转移中的作用机制,长期以来都是一个“黑箱”。
技术瓶颈与突破
要解开上述谜团,就需要能够在单细胞分辨率下,同时分析细胞的基因表达、染色质开放状态(表观遗传调控)以及它们在组织原位的空间位置的技术,单细胞多组学技术(scRNA-seq, snATAC-seq) 与空间转录组学(Stereo-seq让我们能以前所未有的精度,窥见TME的全貌和细胞间的“对话”,从而精准定位驱动ESCC转移的核心细胞类型和分子通路。
研究思路
第1阶段:整合多组学,发现关键细胞靶点
收集12例ESCC患者(6例转移/6例非转移)的原发肿瘤及部分癌旁正常组织,进行单细胞RNA测序(scRNA-seq) 和单细胞核ATAC测序(snATAC-seq),对其中5例患者的组织进行高分辨率空间转录组测序(Stereo-seq)。通过细胞聚类、细胞通讯分析(CellChat)和空间邻近性计算,并结合免疫荧光、流式细胞术及大样本组织芯片进行临床验证,锁定了在转移灶中特异性富集、信号输出活跃且与肿瘤细胞空间邻近的 GPR116+周细胞亚群。
第2阶段:细胞亚群分化机制解析
通过 SCENIC单细胞调控网络分析 从多组学数据中筛选出其特异性激活的转录因子 PRRX1。随后,通过一系列体外功能实验(过表达/敲低)证实PRRX1可直接上调GPR116等标志基因的表达。染色质免疫共沉淀(ChIP) 和报告基因实验则直接证明了PRRX1能结合在GPR116和其下游效应分子EGFL6的启动子区域。最终,在周细胞特异性Prrx1敲除小鼠模型中,观察到肿瘤内GPR116+周细胞的几乎完全缺失,从体内验证了PRRX1的核心驱动作用。
第3阶段:功能机制深度探索
- 促转移机制:通过细胞互作分析、免疫共沉淀 验证了GPR116+周细胞通过分泌EGFL6与肿瘤细胞表面的整合素β1结合。后续的体外侵袭实验、小鼠转移模型及信号通路抑制剂挽救实验共同证实,该互作通过激活NF-κB通路促进上皮-间质转化(EMT)和转移。
- 免疫抑制机制:通过多重免疫荧光、体外共培养模型和流式细胞术,发现GPR116+周细胞能诱导调节性T细胞(Treg)扩增、促使CD8+ T细胞功能耗竭,并通过integrin β1信号上调肿瘤细胞PD-L1表达,从而协同抑制抗肿瘤免疫。
第4阶段:临床转化
通过大样本组织微阵列和血清ELISA检测,验证了血清EGFL6作为无创诊断和预后生物标志物的巨大潜力。在皮下移植瘤和自发成瘤两种小鼠模型中,验证了靶向integrin β1(使用抑制剂ATN-161或抗体Volociximab)与抗PD-1免疫疗法的联合方案,能协同抑制肿瘤生长和远处转移,且显示出良好的安全性。
主要研究结果
通过整合单细胞多组学与空间转录组数据,本研究首次在食管鳞癌(ESCC)肿瘤微环境中鉴定出一个特征鲜明的周细胞新亚群——GPR116+周细胞。该亚群在发生转移的肿瘤组织中显著富集,其丰度与患者的不良预后密切相关,被确立为一个独立的预后风险因素。
2. 揭示其上游分化调控机制:由PRRX1转录因子驱动
研究发现,GPR116+周细胞的分化并非随机,而是受转录因子PRRX1的精确调控。通过生物信息学分析和基因敲除小鼠模型验证,证实PRRX1能直接结合到GPR116和EGFL6的启动子区域,激活其表达,是形成该促转移亚群的“总开关”。
3. 阐明其促进转移的核心分子通路:EGFL6-Integrin β1-NF-κB轴
在机制上,GPR116+周细胞通过分泌信号蛋白EGFL6,与肿瘤细胞表面的受体整合素β1(ITGB1)结合,进而激活下游的NF-κB信号通路。这条分子轴的激活,直接增强了肿瘤细胞的侵袭能力和上皮-间质转化(EMT)进程,从而驱动癌症转移。
4. 发现其强大的免疫抑制功能
除了直接促转移,GPR116+周细胞还扮演着“免疫破坏者”的角色。它们能够诱导CD8+ T细胞功能耗竭、促进调节性T细胞(Treg)的扩增,并上调肿瘤细胞表面的免疫检查点蛋白PD-L1的表达,共同塑造了一个抑制性的肿瘤免疫微环境,解释了其导致免疫治疗耐药的原因。
5. 提出潜在的临床转化应用
研究证实,分泌蛋白EGFL6在患者血清中含量显著升高,可作为诊断和预测转移的无创生物标志物。更重要的是,在动物模型中,使用靶向整合素β1的抗体(Volociximab)或小分子抑制剂(ATN-161),能够有效抑制肿瘤转移,并且与抗PD-1免疫疗法展现出强大的协同效应,为克服晚期食管鳞癌的转移与免疫耐药提供了极具前景的联合治疗新策略。
● Fig. 1a-c:研究设计及细胞通讯分析显示,周细胞是转移性ESCC中信号输出最强的细胞类型,与上皮细胞相互作用显著增强。
● Fig. 1d-e:空间分析显示转移灶中周细胞与肿瘤细胞空间距离最近。
● Fig. 1f-h:周细胞分群鉴定出GPR116+亚群,在转移患者中比例显著升高。
● Fig. 1i-j:Stereo-seq空间分布显示GPR116+周细胞在转移患者肿瘤区域富集。
● Fig. 1k-m:免疫荧光和电镜确认周细胞身份,RT-qPCR验证GPR116+亚群标志基因表达。
● Fig. 1n-o:流式细胞术和组织芯片显示GPR116+周细胞在转移患者中显著增加。
● Fig. 1p-q:生存分析显示GPR116+周细胞高比例与患者不良预后独立相关。
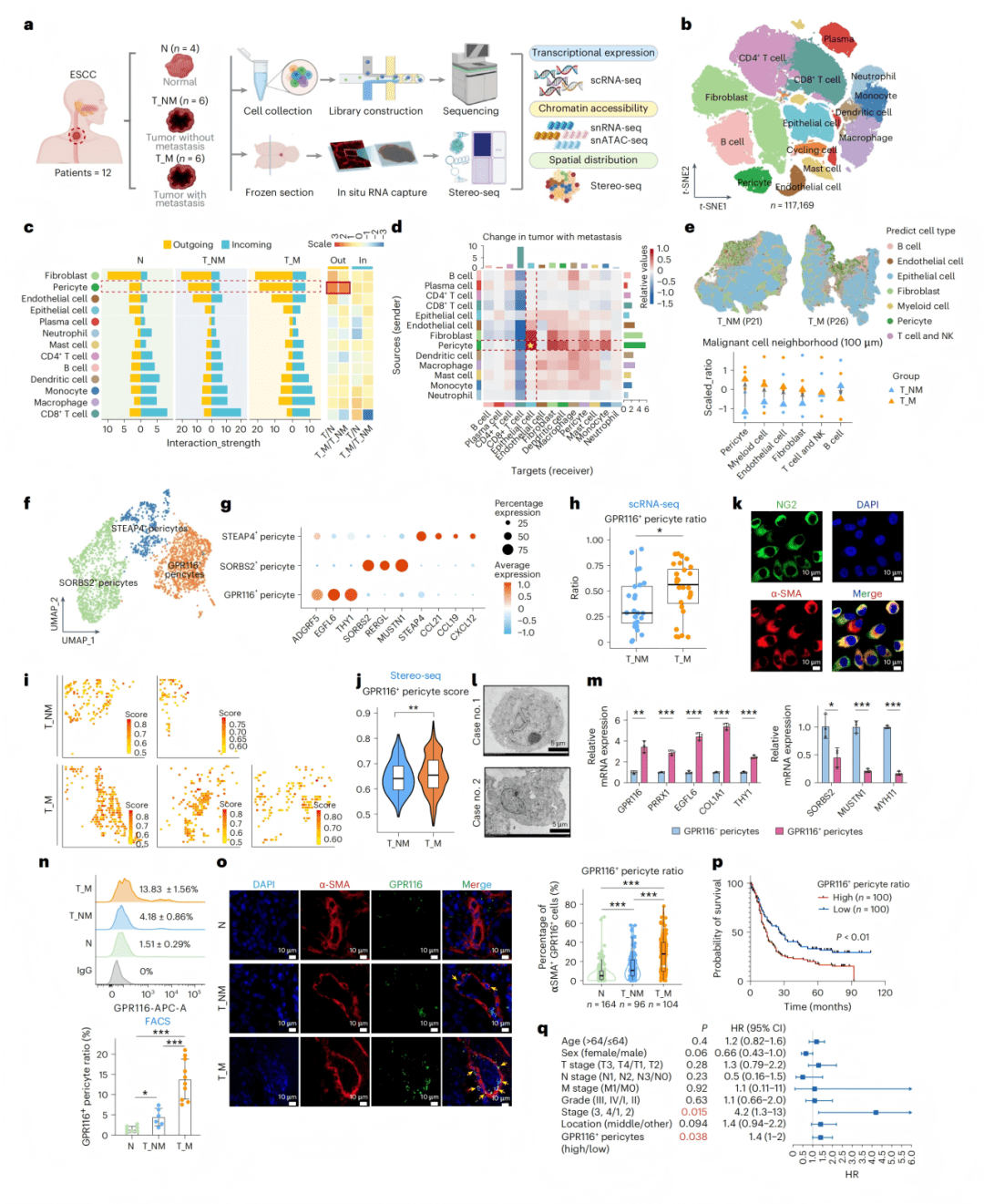
● Fig. 2a-c:SCENIC分析筛选出PRRX1是驱动GPR116+周细胞分化的关键转录因子。
● Fig. 2d-e:UMAP可视化显示PRRX1在GPR116+周细胞中特异性高表达。
● Fig. 2f:Western blot验证PRRX1在肿瘤组织和GPR116+周细胞中高表达。
● Fig. 2g:流式细胞术显示PRRX1过表达显著增加GPR116+细胞比例。
● Fig. 2h:snATAC-seq显示GPR116基因座在GPR116+周细胞中染色质可及性更高。
● Fig. 2i-j:ChIP实验证明PRRX1直接结合GPR116和EGFL6启动子区域。
● Fig. 2k-l:周细胞特异性Prrx1敲除小鼠中GPR116+细胞几乎完全缺失。
● Fig. 3a-c:细胞互作分析显示GPR116+周细胞与高EMT潜能肿瘤细胞相互作用最强。
● Fig. 3d-e:空间分析显示GPR116+周细胞在肿瘤侵袭前沿富集。
● Fig. 3f:3D侵袭实验显示GPR116+周细胞显著增强肿瘤细胞侵袭能力。
● Fig. 3g-h:尾静脉肺转移模型显示GPR116+周细胞促进肺转移。
● Fig. 3i-j:足垫淋巴转移模型显示GPR116+周细胞促进淋巴结转移。
● Fig. 3k-m:Prrx1敲除小鼠模型中淋巴结转移率显著降低。

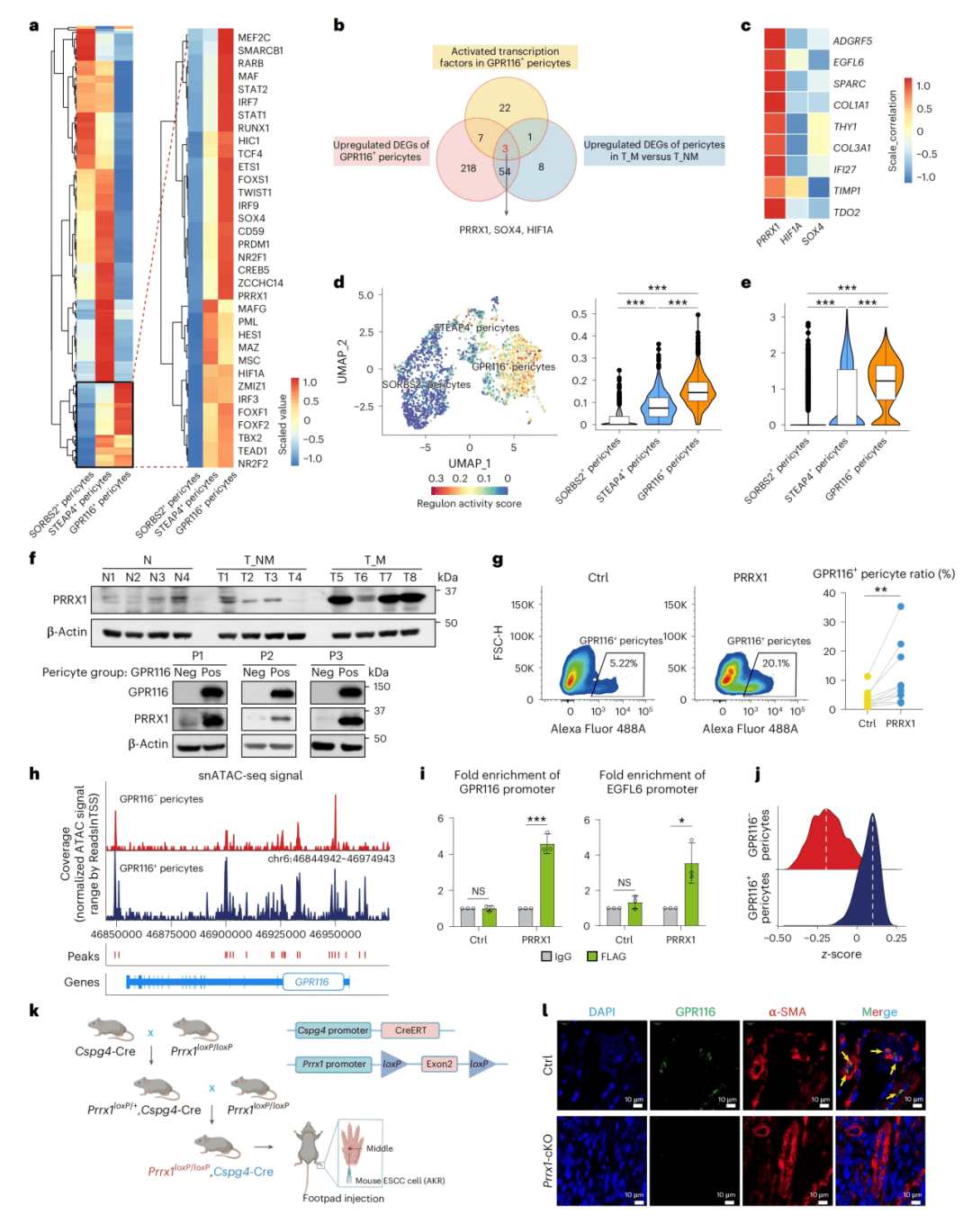
● Fig. 4a-b:差异基因分析显示EGFL6是GPR116+周细胞中最上调的分泌蛋白。
● Fig. 4c:EGFL6在周细胞中特异性高表达。
● Fig. 4d:Prrx1敲除小鼠中EGFL6表达显著降低。
● Fig. 4e-g:EGFL6在转移患者中高表达,且与GPR116+周细胞特征正相关。
● Fig. 4h-i:体外侵袭实验显示EGFL6剂量依赖性增强肿瘤细胞侵袭。
● Fig. 4j:Western blot显示EGFL6诱导EMT相关蛋白表达。
● Fig. 4k-m:组织芯片显示EGFL6高表达与不良预后相关。
● Fig. 4n-r:血清ELISA显示EGFL6是有效的诊断和预后生物标志物。
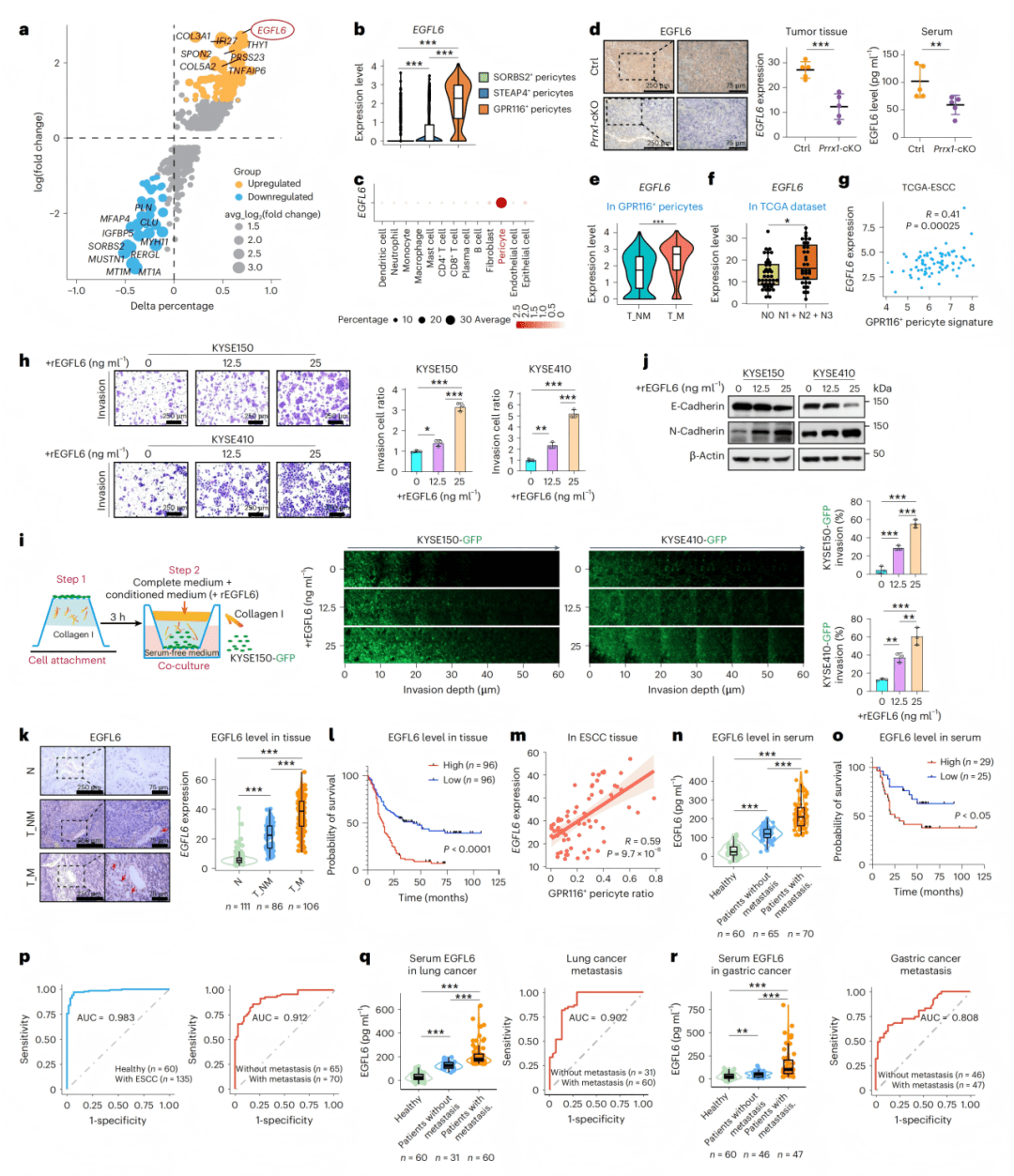
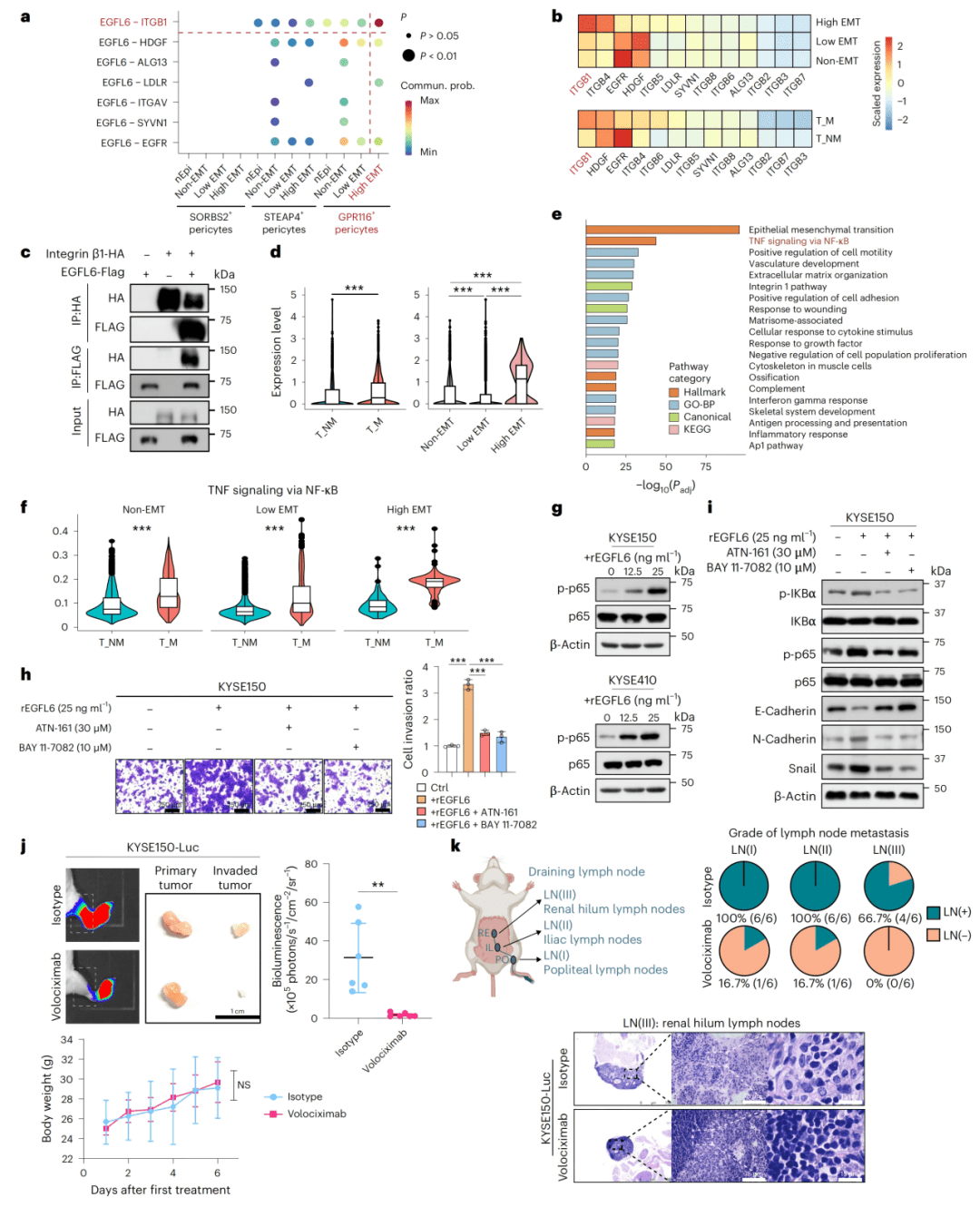
● Fig. 5a-b:细胞互作显示EGFL6-integrin β1是高EMT肿瘤细胞的主要作用对。
● Fig. 5c:Co-IP证实EGFL6与integrin β1直接结合。
● Fig. 5d:Integrin β1在高EMT肿瘤细胞和转移患者中高表达。
● Fig. 5e-f:通路分析显示NF-κB通路在转移组和高EMT细胞中激活。
● Fig. 5g:Western blot显示EGFL6剂量依赖性激活NF-κB通路。
● Fig. 5h-i:抑制剂实验显示阻断integrin β1或NF-κB逆转EGFL6的促转移作用。
● Fig. 5j-k:Volociximab治疗显著抑制淋巴转移。
● Fig. 6a:多重免疫荧光显示GPR116+周细胞与PD-1+CD8+T细胞和Treg空间共定位。
● Fig. 6b-c:共培养实验显示GPR116+周细胞诱导T细胞耗竭和Treg扩增。
● Fig. 6d:肿瘤杀伤实验显示GPR116+周细胞削弱免疫细胞杀伤功能。
● Fig. 6e:流式细胞术显示GPR116+周细胞降低CD8+T细胞毒性分子分泌。
● Fig. 6f-g:GPR116+周细胞通过integrin β1信号上调肿瘤细胞PD-L1表达。
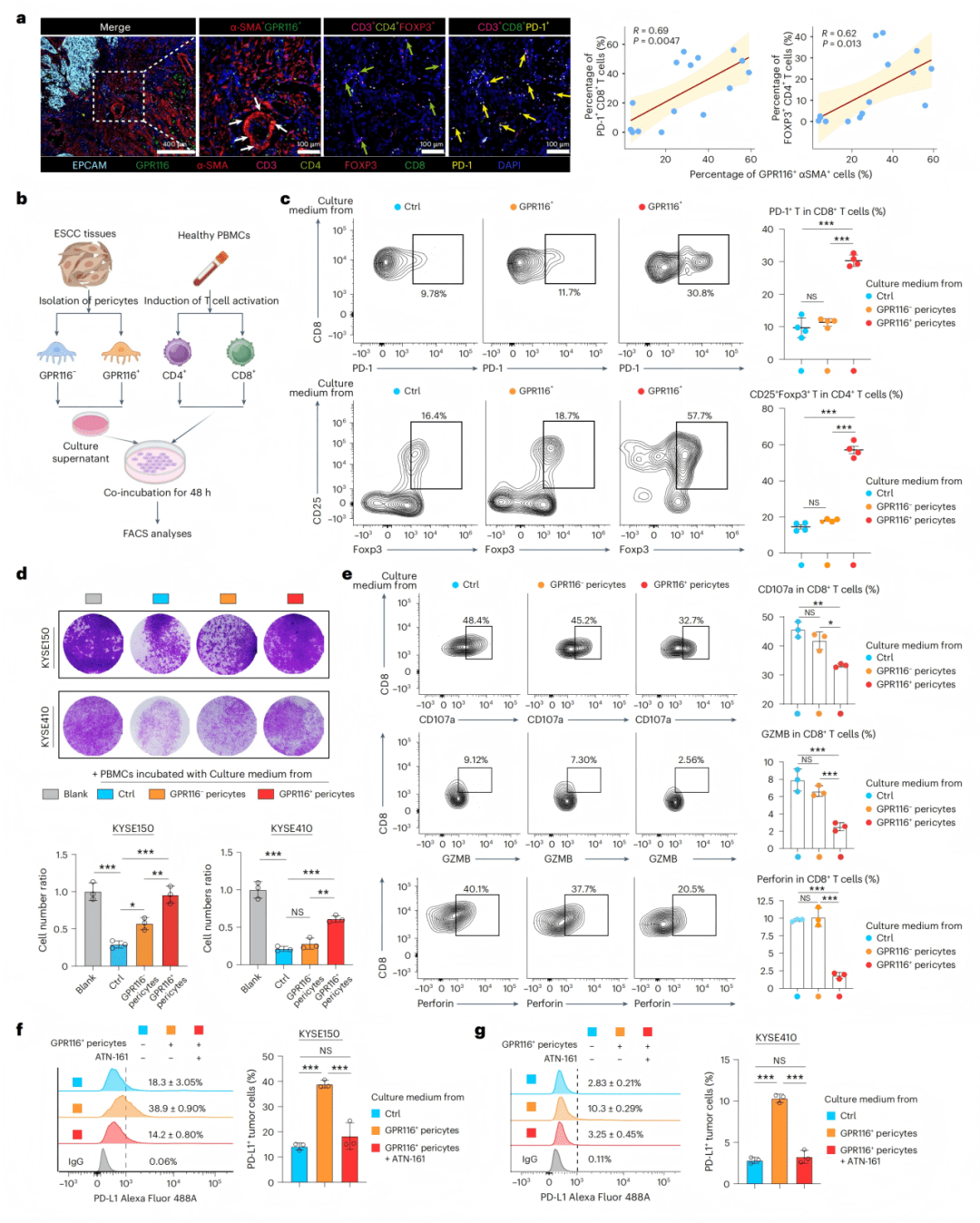
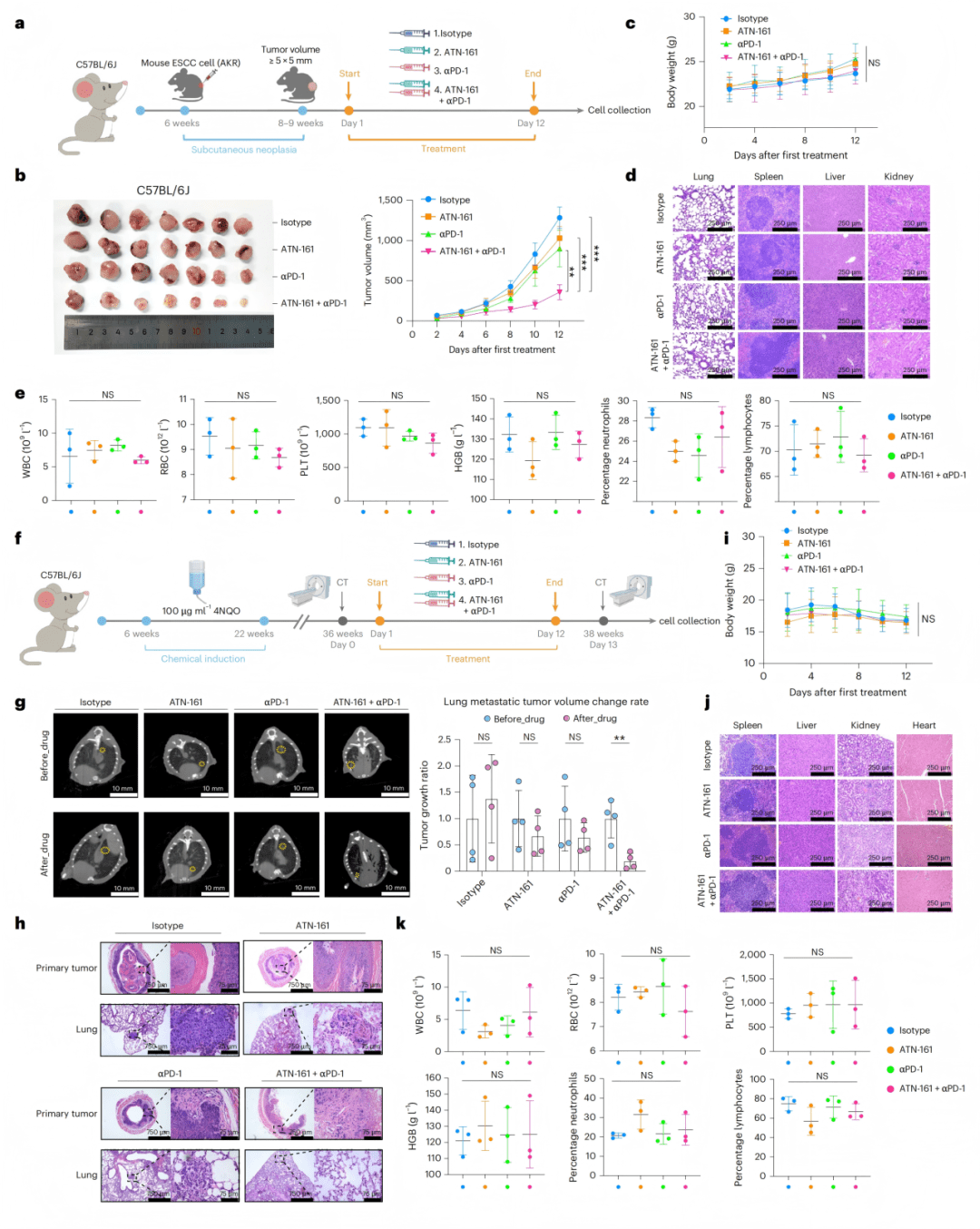
● Fig. 7a-e:皮下移植瘤模型显示ATN-161与αPD-1联用显著抑制肿瘤生长,且安全性良好。
● Fig. 7f-k:自发肿瘤模型显示联合治疗显著抑制原发瘤进展和肺转移,未出现明显毒性反应。
Pei X, Liu Z, Tang L, et al. Single-cell multi-omic and spatial profiling of esophageal squamous cell carcinoma reveals the immunosuppressive role of GPR116+ pericytes in cancer metastasis. Nat Genet. 2025;57(10):2494-2508. doi:10.1038/s41588-025-02341-9